Améliorer l'estimation d'énergie d'un Hamiltonien de chimie avec SQD
Dans ce tutoriel, on implémente un patron Qiskit montrant comment post-traiter des échantillons quantiques bruités pour trouver une approximation de l'état fondamental d'un Hamiltonien de chimie : la molécule à l'équilibre dans la base 6-31G. On va suivre une approche de diagonalisation quantique basée sur des échantillons pour traiter des échantillons prélevés depuis un ansatz de circuit quantique à 36 qubits (dans ce cas, un circuit LUCJ). Pour tenir compte de l'effet du bruit quantique, la technique de récupération de configuration est utilisée.
Le patron peut être décrit en quatre étapes :
- Étape 1 : Mapper au problème quantique
- Générer un ansatz pour estimer l'état fondamental
- Étape 2 : Optimiser le problème
- Transpiler l'ansatz pour le Backend
- Étape 3 : Exécuter les expériences
- Tirer des échantillons de l'ansatz en utilisant la primitive
Sampler
- Tirer des échantillons de l'ansatz en utilisant la primitive
- Étape 4 : Post-traiter les résultats
- Boucle de récupération de configuration auto-cohérente
- Post-traiter l'ensemble complet des échantillons de chaînes de bits, en utilisant la connaissance a priori du nombre de particules et de l'occupation orbitale moyenne calculée lors de la dernière itération.
- Créer de façon probabiliste des lots de sous-échantillons à partir des chaînes de bits récupérées.
- Projeter et diagonaliser le Hamiltonien moléculaire sur chaque sous-espace échantillonné.
- Sauvegarder l'énergie minimale de l'état fondamental trouvée sur tous les lots et mettre à jour l'occupation orbitale moyenne.
- Boucle de récupération de configuration auto-cohérente
Pour cet exemple, le Hamiltonien d'électrons en interaction prend la forme générique :
/ sont les opérateurs de création/annihilation fermioniques associés au -ième élément de la base et au spin . et sont les intégrales électroniques à un et deux corps.
Le flux de travail SQD avec récupération de configuration auto-cohérente est représenté dans le diagramme suivant.
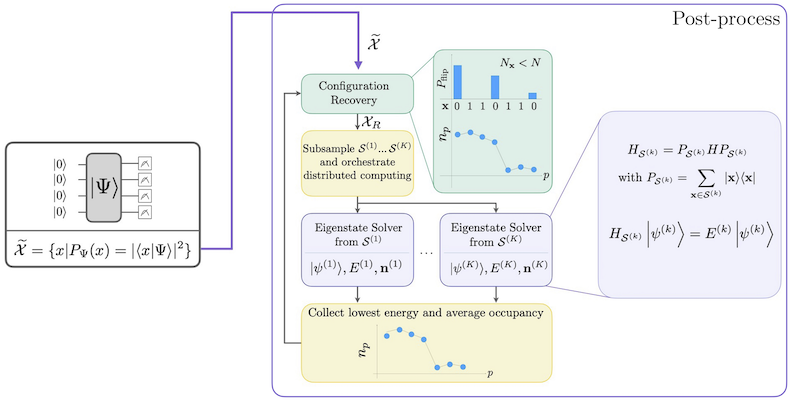
Il est connu que SQD fonctionne bien lorsque l'état propre cible est épars : la fonction d'onde est supportée dans un ensemble d'états de base dont la taille n'augmente pas exponentiellement avec la taille du problème. Dans ce scénario, la diagonalisation du Hamiltonien projeté dans le sous-espace défini par :
donne une bonne approximation de l'état propre cible. Le rôle du dispositif quantique est de produire des échantillons des membres de uniquement. D'abord, un circuit quantique prépare l'état dans le dispositif quantique. L'encodage de Jordan-Wigner est utilisé. Par conséquent, les membres de la base computationnelle représentent des états de Fock (configurations/déterminants électroniques). Le circuit est échantillonné dans la base computationnelle, ce qui donne l'ensemble des configurations bruitées . Les configurations sont représentées par des chaînes de bits. L'ensemble est ensuite transmis au bloc de post-traitement classique, où la technique de récupération de configuration auto-cohérente est utilisée. Dans le cadre SQD, le rôle du dispositif quantique est de produire une distribution de probabilité.
Étape 1 : Mapper le problème à un circuit quantique
Dans ce tutoriel, on va approximer l'énergie de l'état fondamental d'une molécule . D'abord, on va spécifier la molécule et ses propriétés. Ensuite, on va créer un ansatz local unitary cluster Jastrow (LUCJ) (circuit quantique) pour générer des échantillons depuis un ordinateur quantique afin d'estimer l'énergie de l'état fondamental.
D'abord, on va spécifier la molécule et ses propriétés.
# Added by doQumentation — required packages for this notebook
!pip install -q ffsim matplotlib numpy pyscf qiskit qiskit-addon-sqd qiskit-ibm-runtime
import warnings
warnings.filterwarnings("ignore")
import pyscf
import pyscf.cc
import pyscf.mcscf
# Specify molecule properties
open_shell = False
spin_sq = 0
# Build N2 molecule
mol = pyscf.gto.Mole()
mol.build(
atom=[["N", (0, 0, 0)], ["N", (1.0, 0, 0)]],
basis="6-31g",
symmetry="Dooh",
)
# Define active space
n_frozen = 2
active_space = range(n_frozen, mol.nao_nr())
# Get molecular integrals
scf = pyscf.scf.RHF(mol).run()
num_orbitals = len(active_space)
n_electrons = int(sum(scf.mo_occ[active_space]))
num_elec_a = (n_electrons + mol.spin) // 2
num_elec_b = (n_electrons - mol.spin) // 2
cas = pyscf.mcscf.CASCI(scf, num_orbitals, (num_elec_a, num_elec_b))
mo = cas.sort_mo(active_space, base=0)
hcore, nuclear_repulsion_energy = cas.get_h1cas(mo)
eri = pyscf.ao2mo.restore(1, cas.get_h2cas(mo), num_orbitals)
# Compute exact energy
exact_energy = cas.run().e_tot
converged SCF energy = -108.835236570775
CASCI E = -109.046671778080 E(CI) = -32.8155692383188 S^2 = 0.0000000
Ensuite, on va créer l'ansatz. L'ansatz LUCJ est un circuit quantique paramétré, et on va l'initialiser avec les amplitudes t2 et t1 obtenues depuis un calcul CCSD.
# Get CCSD t2 amplitudes for initializing the ansatz
ccsd = pyscf.cc.CCSD(scf, frozen=[i for i in range(mol.nao_nr()) if i not in active_space]).run()
t1 = ccsd.t1
t2 = ccsd.t2
E(CCSD) = -109.0398256929734 E_corr = -0.2045891221988311
On va utiliser le package ffsim pour créer et initialiser l'ansatz avec les amplitudes t2 et t1 calculées ci-dessus. Comme notre molécule a un état de Hartree-Fock à couches fermées, on va utiliser la variante équilibrée en spin de l'ansatz UCJ, UCJOpSpinBalanced.
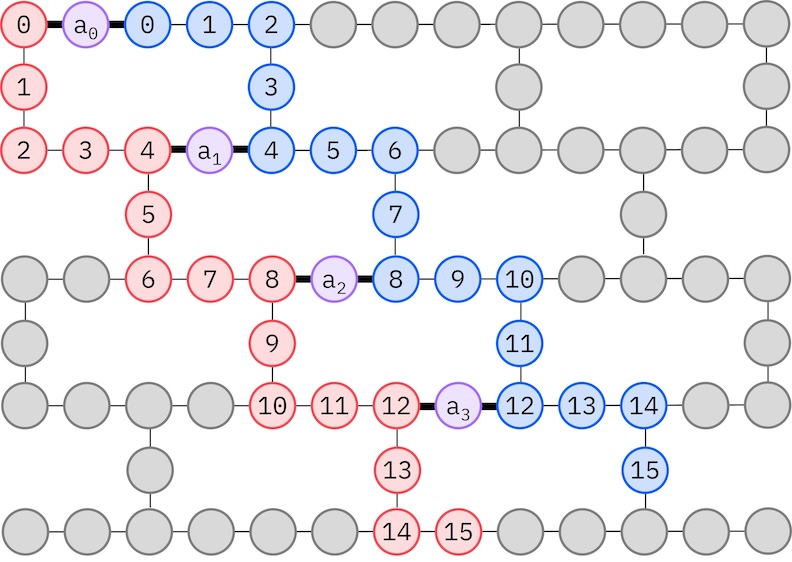
Comme notre matériel IBM cible a une topologie heavy-hex, on va adopter le motif zig-zag pour les interactions de qubit. Dans ce motif, les orbitales (représentées par des qubits) de même spin sont connectées avec une topologie en ligne (cercles rouges et bleus) où chaque ligne prend une forme en zig-zag en raison de la connectivité heavy-hex du matériel cible. De plus, en raison de la topologie heavy-hex, les orbitales de spins différents ont des connexions entre chaque 4e orbitale (0, 4, 8, etc.) (cercles violets).
import ffsim
from qiskit import QuantumCircuit, QuantumRegister
n_reps = 1
alpha_alpha_indices = [(p, p + 1) for p in range(num_orbitals - 1)]
alpha_beta_indices = [(p, p) for p in range(0, num_orbitals, 4)]
ucj_op = ffsim.UCJOpSpinBalanced.from_t_amplitudes(
t2=t2,
t1=t1,
n_reps=n_reps,
interaction_pairs=(alpha_alpha_indices, alpha_beta_indices),
)
nelec = (num_elec_a, num_elec_b)
# create an empty quantum circuit
qubits = QuantumRegister(2 * num_orbitals, name="q")
circuit = QuantumCircuit(qubits)
# prepare Hartree-Fock state as the reference state and append it to the quantum circuit
circuit.append(ffsim.qiskit.PrepareHartreeFockJW(num_orbitals, nelec), qubits)
# apply the UCJ operator to the reference state
circuit.append(ffsim.qiskit.UCJOpSpinBalancedJW(ucj_op), qubits)
circuit.measure_all()
Étape 2 : Optimiser le problème
Ensuite, on va optimiser notre circuit pour un matériel cible. On doit choisir le dispositif matériel à utiliser avant d'optimiser notre circuit. On va utiliser un faux Backend de 127 qubits depuis qiskit_ibm_runtime pour émuler un vrai dispositif. Pour exécuter sur un vrai QPU, il suffit de remplacer le faux Backend par un vrai Backend. Consulte la documentation Qiskit IBM Runtime pour plus d'informations.
from qiskit_ibm_runtime.fake_provider import FakeSherbrooke
backend = FakeSherbrooke()
Ensuite, on recommande les étapes suivantes pour optimiser l'ansatz et le rendre compatible avec le matériel.
- Sélectionner des qubits physiques (
initial_layout) depuis le matériel cible qui respecte le motif zig-zag décrit ci-dessus. Disposer les qubits dans ce motif conduit à un circuit efficace compatible avec le matériel, avec moins de portes. - Générer un gestionnaire de passes par étapes en utilisant la fonction generate_preset_pass_manager de Qiskit avec ton choix de
backendetinitial_layout. - Définir l'étape
pre_initde ton gestionnaire de passes par étapes àffsim.qiskit.PRE_INIT.ffsim.qiskit.PRE_INITinclut des passes du Transpiler Qiskit qui décomposent les portes en rotations orbitales puis fusionnent les rotations orbitales, ce qui donne moins de portes dans le circuit final. - Exécuter le gestionnaire de passes sur ton circuit.
from qiskit.transpiler.preset_passmanagers import generate_preset_pass_manager
spin_a_layout = [0, 14, 18, 19, 20, 33, 39, 40, 41, 53, 60, 61, 62, 72, 81, 82]
spin_b_layout = [2, 3, 4, 15, 22, 23, 24, 34, 43, 44, 45, 54, 64, 65, 66, 73]
initial_layout = spin_a_layout + spin_b_layout
pass_manager = generate_preset_pass_manager(
optimization_level=3, backend=backend, initial_layout=initial_layout
)
# without PRE_INIT passes
isa_circuit = pass_manager.run(circuit)
print(f"Gate counts (w/o pre-init passes): {isa_circuit.count_ops()}")
# with PRE_INIT passes
# We will use the circuit generated by this pass manager for hardware execution
pass_manager.pre_init = ffsim.qiskit.PRE_INIT
isa_circuit = pass_manager.run(circuit)
print(f"Gate counts (w/ pre-init passes): {isa_circuit.count_ops()}")
Gate counts (w/o pre-init passes): OrderedDict({'rz': 4420, 'sx': 3432, 'ecr': 1366, 'x': 239, 'measure': 32, 'barrier': 1})
Gate counts (w/ pre-init passes): OrderedDict({'rz': 2460, 'sx': 2156, 'ecr': 730, 'x': 71, 'measure': 32, 'barrier': 1})
Étape 3 : Exécuter les expériences
Après avoir optimisé le circuit pour l'exécution matérielle, on est prêt à l'exécuter sur le matériel cible et à collecter des échantillons pour l'estimation de l'énergie de l'état fondamental. Comme on n'a qu'un seul circuit, on va utiliser le mode d'exécution par Job de Qiskit Runtime et exécuter notre circuit.
Note : On a commenté le code pour exécuter le circuit sur un QPU et on l'a laissé comme référence pour l'utilisateur. Au lieu d'exécuter sur du vrai matériel dans ce guide, on va simplement générer des échantillons aléatoires tirés de la distribution uniforme.
import numpy as np
from qiskit_addon_sqd.counts import generate_bit_array_uniform
# from qiskit_ibm_runtime import SamplerV2 as Sampler
# sampler = Sampler(mode=backend)
# job = sampler.run([isa_circuit], shots=10_000)
# primitive_result = job.result()
# pub_result = primitive_result[0]
# bit_array = pub_result.data.meas
rng = np.random.default_rng(24)
bit_array = generate_bit_array_uniform(10_000, num_orbitals * 2, rand_seed=rng)
Étape 4 : Post-traiter les résultats
Maintenant, on exécute l'algorithme SQD en utilisant la fonction diagonalize_fermionic_hamiltonian. Consulte la documentation API pour les explications des arguments de cette fonction.
Le solveur inclus dans l'addon SQD utilise l'implémentation de CI sélectionné de PySCF, spécifiquement pyscf.fci.selected_ci.kernel_fixed_space. L'exemple ci-dessous montre également comment passer des arguments nommés à cette fonction via le solveur inclus. Ici on passe l'argument max_cycle.
from functools import partial
from qiskit_addon_sqd.fermion import SCIResult, diagonalize_fermionic_hamiltonian, solve_sci_batch
# SQD options
energy_tol = 1e-3
occupancies_tol = 1e-3
max_iterations = 5
# Eigenstate solver options
num_batches = 1
samples_per_batch = 300
symmetrize_spin = True
carryover_threshold = 1e-4
max_cycle = 200
# Pass options to the built-in eigensolver. If you just want to use the defaults,
# you can omit this step, in which case you would not specify the sci_solver argument
# in the call to diagonalize_fermionic_hamiltonian below.
sci_solver = partial(solve_sci_batch, spin_sq=0.0, max_cycle=max_cycle)
# List to capture intermediate results
result_history = []
def callback(results: list[SCIResult]):
result_history.append(results)
iteration = len(result_history)
print(f"Iteration {iteration}")
for i, result in enumerate(results):
print(f"\tSubsample {i}")
print(f"\t\tEnergy: {result.energy + nuclear_repulsion_energy}")
print(f"\t\tSubspace dimension: {np.prod(result.sci_state.amplitudes.shape)}")
result = diagonalize_fermionic_hamiltonian(
hcore,
eri,
bit_array,
samples_per_batch=samples_per_batch,
norb=num_orbitals,
nelec=nelec,
num_batches=num_batches,
energy_tol=energy_tol,
occupancies_tol=occupancies_tol,
max_iterations=max_iterations,
sci_solver=sci_solver,
symmetrize_spin=symmetrize_spin,
carryover_threshold=carryover_threshold,
callback=callback,
seed=rng,
)
Iteration 1
Subsample 0
Energy: -105.45358671756313
Subspace dimension: 5476
Iteration 2
Subsample 0
Energy: -107.95172900082163
Subspace dimension: 249001
Iteration 3
Subsample 0
Energy: -108.97460330369815
Subspace dimension: 339889
Iteration 4
Subsample 0
Energy: -109.02739376648793
Subspace dimension: 440896
Iteration 5
Subsample 0
Energy: -109.030972328451
Subspace dimension: 597529
Maintenant, on trace les résultats.
Le premier graphe montre qu'après quelques itérations, on estime l'énergie de l'état fondamental à ~16 mH près (la précision chimique est typiquement acceptée à 1 kcal/mol 1.6 mH). Rappelle-toi que les échantillons quantiques dans cette démo étaient du bruit pur. Le signal provient ici de la connaissance a priori de la structure électronique et du Hamiltonien moléculaire.
Le second graphe montre l'occupation moyenne de chaque orbitale spatiale après la dernière itération. On peut voir que les électrons spin-haut et spin-bas occupent les cinq premières orbitales avec une haute probabilité dans nos solutions.
import matplotlib.pyplot as plt
# Data for energies plot
x1 = range(len(result_history))
min_e = [
min(result, key=lambda res: res.energy).energy + nuclear_repulsion_energy
for result in result_history
]
e_diff = [abs(e - exact_energy) for e in min_e]
yt1 = [1.0, 1e-1, 1e-2, 1e-3, 1e-4]
# Chemical accuracy (+/- 1 milli-Hartree)
chem_accuracy = 0.001
# Data for avg spatial orbital occupancy
y2 = np.sum(result.orbital_occupancies, axis=0)
x2 = range(len(y2))
fig, axs = plt.subplots(1, 2, figsize=(12, 6))
# Plot energies
axs[0].plot(x1, e_diff, label="energy error", marker="o")
axs[0].set_xticks(x1)
axs[0].set_xticklabels(x1)
axs[0].set_yticks(yt1)
axs[0].set_yticklabels(yt1)
axs[0].set_yscale("log")
axs[0].set_ylim(1e-4)
axs[0].axhline(y=chem_accuracy, color="#BF5700", linestyle="--", label="chemical accuracy")
axs[0].set_title("Approximated Ground State Energy Error vs SQD Iterations")
axs[0].set_xlabel("Iteration Index", fontdict={"fontsize": 12})
axs[0].set_ylabel("Energy Error (Ha)", fontdict={"fontsize": 12})
axs[0].legend()
# Plot orbital occupancy
axs[1].bar(x2, y2, width=0.8)
axs[1].set_xticks(x2)
axs[1].set_xticklabels(x2)
axs[1].set_title("Avg Occupancy per Spatial Orbital")
axs[1].set_xlabel("Orbital Index", fontdict={"fontsize": 12})
axs[1].set_ylabel("Avg Occupancy", fontdict={"fontsize": 12})
print(f"Exact energy: {exact_energy:.5f} Ha")
print(f"SQD energy: {min_e[-1]:.5f} Ha")
print(f"Absolute error: {e_diff[-1]:.5f} Ha")
plt.tight_layout()
plt.show()
Exact energy: -109.04667 Ha
SQD energy: -109.03097 Ha
Absolute error: 0.01570 Ha
