Diagonalisation quantique de Krylov basée sur les échantillons d'un modèle de réseau fermionique
Estimation d'utilisation : neuf secondes sur un processeur Heron r2 (REMARQUE : il s'agit uniquement d'une estimation. Votre temps d'exécution peut varier.)
Résultats d'apprentissage
Après avoir suivi ce tutoriel, les utilisateurs devraient comprendre :
- Comment utiliser l'addon SQD de Qiskit pour approximer l'énergie de l'état fondamental d'un modèle de réseau à l'aide de chaînes de bits échantillonnées depuis une unité de traitement quantique (QPU).
- Comment utiliser ffsim pour construire des circuits d'évolution temporelle pour la simulation fermionique.
- Comment combiner des échantillons provenant de plusieurs circuits pour le post-traitement avec l'algorithme de diagonalisation quantique de Krylov basée sur les échantillons (SQKD).
Prérequis
Nous suggérons aux utilisateurs d'être familiers avec les sujets suivants avant de suivre ce tutoriel :
- Diagonalisation quantique basée sur les échantillons d'un hamiltonien de chimie
- Diagonalisation quantique de Krylov des hamiltoniens de réseau
- Primitives Qiskit
Contexte
Ce tutoriel montre comment utiliser la diagonalisation quantique basée sur les échantillons (SQD) pour estimer l'énergie de l'état fondamental d'un modèle de réseau fermionique. Plus précisément, nous étudions le modèle d'Anderson à impureté unique unidimensionnel (SIAM), utilisé pour décrire les impuretés magnétiques incorporées dans les métaux.
Ce tutoriel suit un flux de travail similaire au tutoriel connexe Diagonalisation quantique basée sur les échantillons d'un hamiltonien de chimie. Cependant, une différence clé réside dans la façon dont les circuits quantiques sont construits. L'autre tutoriel utilise un ansatz variationnel heuristique, ce qui est intéressant pour les hamiltoniens de chimie comportant potentiellement des millions de termes d'interaction. En revanche, ce tutoriel utilise des circuits qui approximent l'évolution temporelle selon le hamiltonien. De tels circuits peuvent être profonds, ce qui rend cette approche plus adaptée aux applications sur des modèles de réseau. Les vecteurs d'état préparés par ces circuits forment la base d'un sous-espace de Krylov, et par conséquent, l'algorithme converge de manière prouvable et efficace vers l'état fondamental, sous des hypothèses appropriées.
L'approche utilisée dans ce tutoriel peut être considérée comme une combinaison des techniques employées dans la SQD et la diagonalisation quantique de Krylov (KQD). L'approche combinée est parfois désignée sous le nom de diagonalisation quantique de Krylov basée sur les échantillons (SQKD). Consulte Diagonalisation quantique de Krylov des hamiltoniens de réseau pour un tutoriel sur la méthode KQD.
Ce tutoriel est basé sur les travaux "Quantum-Centric Algorithm for Sample-Based Krylov Diagonalization", auxquels tu peux te référer pour plus de détails.
Modèle d'Anderson à impureté unique (SIAM)
Le hamiltonien SIAM unidimensionnel est une somme de trois termes :
où
Ici, sont les opérateurs de création/annihilation fermioniques pour le site du bain avec le spin , sont les opérateurs de création/annihilation pour le mode d'impureté, et . , et sont des nombres réels décrivant les interactions de saut, sur site et d'hybridation, et est un nombre réel spécifiant le potentiel chimique.
Note que le hamiltonien est une instance spécifique du hamiltonien générique d'électrons en interaction,
où est constitué de termes à un corps, qui sont quadratiques en opérateurs de création et d'annihilation fermioniques, et est constitué de termes à deux corps, qui sont quartiques. Pour le SIAM,
et contient le reste des termes du hamiltonien. Afin de représenter le hamiltonien de manière programmatique, nous stockons la matrice et le tenseur .
Bases de position et d'impulsion
En raison de la symétrie de translation approximative dans , nous ne nous attendons pas à ce que l'état fondamental soit creux dans la base de position (la base orbitale dans laquelle le hamiltonien est spécifié ci-dessus). Les performances de la SQD ne sont garanties que si l'état fondamental est creux, c'est-à-dire qu'il a un poids significatif sur seulement un petit nombre d'états de base computationnels. Pour améliorer la parcimonie de l'état fondamental, nous effectuons la simulation dans la base orbitale dans laquelle est diagonal. Nous appelons cette base la base d'impulsion. Comme est un hamiltonien fermionique quadratique, il peut être efficacement diagonalisé par une rotation orbitale.
Évolution temporelle approximative selon le hamiltonien
Pour approximer l'évolution temporelle selon le hamiltonien, nous utilisons une décomposition de Trotter-Suzuki du second ordre,
Sous la transformation de Jordan-Wigner, l'évolution temporelle selon revient à une seule porte CPhase entre les orbitales de spin-up et spin-down au site d'impureté. Comme est un hamiltonien fermionique quadratique, l'évolution temporelle selon revient à une rotation orbitale.
Les états de base de Krylov , où est la dimension du sous-espace de Krylov, sont formés par application répétée d'un seul pas de Trotter, de sorte que
Dans le flux de travail SQD suivant, nous échantillonnerons à partir de cet ensemble de circuits et post-traiterons l'ensemble combiné de chaînes de bits avec la SQD. Cette approche contraste avec celle utilisée dans le tutoriel connexe Diagonalisation quantique basée sur les échantillons d'un hamiltonien de chimie, où les échantillons étaient tirés d'un seul circuit variationnel heuristique.
Prérequis
Avant de commencer ce tutoriel, assure-toi d'avoir installé les éléments suivants :
- Qiskit SDK v1.0 ou ultérieur, avec le support de visualisation
- Qiskit Runtime v0.22 ou ultérieur (
pip install qiskit-ibm-runtime) - Module complémentaire SQD Qiskit v0.11 ou ultérieur (
pip install qiskit-addon-sqd) - ffsim v0.0.72 ou ultérieur (
pip install ffsim)
Exemple à petite échelle avec simulateur
Étape 1 : Associer le problème à un circuit quantique
Tout d'abord, nous générons le hamiltonien SIAM dans la base de position. Le hamiltonien est représenté par la matrice et le tenseur . Ensuite, nous le faisons pivoter vers la base d'impulsion. Dans la base de position, nous plaçons l'impureté au premier site. Cependant, lorsque nous passons à la base d'impulsion, nous déplaçons l'impureté vers un site central pour faciliter les interactions avec les autres orbitales.
# Added by doQumentation — required packages for this notebook
!pip install -q ffsim matplotlib numpy pyscf qiskit qiskit-addon-sqd qiskit-ibm-runtime scipy
import numpy as np
import pyscf.fci
def siam_hamiltonian(
norb: int,
hopping: float,
onsite: float,
hybridization: float,
chemical_potential: float,
) -> tuple[np.ndarray, np.ndarray]:
"""Hamiltonian for the single-impurity Anderson model."""
# Place the impurity on the first site
impurity_orb = 0
# One body matrix elements in the "position" basis
h1e = np.zeros((norb, norb))
np.fill_diagonal(h1e[:, 1:], -hopping)
np.fill_diagonal(h1e[1:, :], -hopping)
h1e[impurity_orb, impurity_orb + 1] = -hybridization
h1e[impurity_orb + 1, impurity_orb] = -hybridization
h1e[impurity_orb, impurity_orb] = chemical_potential
# Two body matrix elements in the "position" basis
h2e = np.zeros((norb, norb, norb, norb))
h2e[impurity_orb, impurity_orb, impurity_orb, impurity_orb] = onsite
return h1e, h2e
def momentum_basis(norb: int) -> np.ndarray:
"""Get the orbital rotation to change from the position to the momentum basis."""
n_bath = norb - 1
# Orbital rotation that diagonalizes the bath (non-interacting system)
hopping_matrix = np.zeros((n_bath, n_bath))
np.fill_diagonal(hopping_matrix[:, 1:], -1)
np.fill_diagonal(hopping_matrix[1:, :], -1)
_, vecs = np.linalg.eigh(hopping_matrix)
# Expand to include impurity
orbital_rotation = np.zeros((norb, norb))
# Impurity is on the first site
orbital_rotation[0, 0] = 1
orbital_rotation[1:, 1:] = vecs
# Move the impurity to the center
new_index = n_bath // 2
perm = np.r_[1 : (new_index + 1), 0, (new_index + 1) : norb]
orbital_rotation = orbital_rotation[:, perm]
return orbital_rotation
def rotated(
h1e: np.ndarray, h2e: np.ndarray, orbital_rotation: np.ndarray
) -> tuple[np.ndarray, np.ndarray]:
"""Rotate the orbital basis of a Hamiltonian."""
h1e_rotated = np.einsum(
"ab,Aa,Bb->AB",
h1e,
orbital_rotation,
orbital_rotation.conj(),
optimize="greedy",
)
h2e_rotated = np.einsum(
"abcd,Aa,Bb,Cc,Dd->ABCD",
h2e,
orbital_rotation,
orbital_rotation.conj(),
orbital_rotation,
orbital_rotation.conj(),
optimize="greedy",
)
return h1e_rotated, h2e_rotated
# Total number of spatial orbitals, including the bath sites and the impurity
# This should be an even number
norb = 8
# System is half-filled
nelec = (norb // 2, norb // 2)
# One orbital is the impurity, the rest are bath sites
n_bath = norb - 1
# Hamiltonian parameters
hybridization = 1.0
hopping = 1.0
onsite = 10.0
chemical_potential = -0.5 * onsite
# Generate Hamiltonian in position basis
h1e, h2e = siam_hamiltonian(
norb=norb,
hopping=hopping,
onsite=onsite,
hybridization=hybridization,
chemical_potential=chemical_potential,
)
# Rotate to momentum basis
orbital_rotation = momentum_basis(norb)
h1e_momentum, h2e_momentum = rotated(h1e, h2e, orbital_rotation.T.conj())
# In the momentum basis, the impurity is placed in the center
impurity_index = n_bath // 2
# Use PySCF to compute the exact ground state energy
reference_energy, _ = pyscf.fci.direct_spin1.kernel(h1e, h2e, norb, nelec)
from typing import Sequence
import ffsim
import scipy
from qiskit import QuantumCircuit, QuantumRegister
from qiskit.circuit import CircuitInstruction, Qubit
from qiskit.circuit.library import CPhaseGate, XGate, XXPlusYYGate
def prepare_initial_state(qubits: Sequence[Qubit], norb: int, nocc: int):
"""Prepare initial state."""
assert norb >= 8
x_gate = XGate()
rot = XXPlusYYGate(0.5 * np.pi, -0.5 * np.pi)
for i in range(nocc):
yield CircuitInstruction(x_gate, [qubits[i]])
yield CircuitInstruction(x_gate, [qubits[norb + i]])
for i in range(3):
for j in range(nocc - i - 1, nocc + i, 2):
yield CircuitInstruction(rot, [qubits[j], qubits[j + 1]])
yield CircuitInstruction(
rot, [qubits[norb + j], qubits[norb + j + 1]]
)
yield CircuitInstruction(rot, [qubits[j + 1], qubits[j + 2]])
yield CircuitInstruction(
rot, [qubits[norb + j + 1], qubits[norb + j + 2]]
)
def trotter_step(
qubits: Sequence[Qubit],
time_step: float,
one_body_evolution: np.ndarray,
h2e: np.ndarray,
impurity_index: int,
norb: int,
):
"""A Trotter step."""
# Assume the two-body interaction is just the on-site interaction of the impurity
onsite = h2e[
impurity_index, impurity_index, impurity_index, impurity_index
]
# Two-body evolution for half the time
yield CircuitInstruction(
CPhaseGate(-0.5 * time_step * onsite),
[qubits[impurity_index], qubits[norb + impurity_index]],
)
# One-body evolution for the full time
yield CircuitInstruction(
ffsim.qiskit.OrbitalRotationJW(norb, one_body_evolution), qubits
)
# Two-body evolution for half the time
yield CircuitInstruction(
CPhaseGate(-0.5 * time_step * onsite),
[qubits[impurity_index], qubits[norb + impurity_index]],
)
# Time step
time_step = 0.2
# Number of Krylov basis states
krylov_dim = 8
# Initialize circuit
qubits = QuantumRegister(2 * norb, name="q")
circuit = QuantumCircuit(qubits)
# Generate initial state
for instruction in prepare_initial_state(qubits, norb=norb, nocc=norb // 2):
circuit.append(instruction)
circuit.measure_all()
# Create list of circuits, starting with the initial state circuit
circuits = [circuit.copy()]
# Add time evolution circuits to the list
one_body_evolution = scipy.linalg.expm(-1j * time_step * h1e_momentum)
for i in range(krylov_dim - 1):
# Remove measurements
circuit.remove_final_measurements()
# Append another Trotter step
for instruction in trotter_step(
qubits,
time_step,
one_body_evolution,
h2e_momentum,
impurity_index,
norb,
):
circuit.append(instruction)
# Measure qubits
circuit.measure_all()
# Add a copy of the circuit to the list
circuits.append(circuit.copy())
Ensuite, nous générons les circuits pour produire les états de base de Krylov. Pour chaque espèce de spin, l'état initial est donné par la superposition de toutes les excitations possibles des trois électrons les plus proches du niveau de Fermi vers les 4 modes vides les plus proches à partir de l'état , et réalisé par l'application de sept portes XXPlusYYGate. Les états évolués dans le temps sont produits par des applications successives d'un pas de Trotter du second ordre.
Pour une description plus détaillée de ce modèle et de la conception des circuits, consultez "Quantum-Centric Algorithm for Sample-Based Krylov Diagonalization".
circuits[0].draw("mpl", scale=0.4, fold=-1)
circuits[-1].draw("mpl", scale=0.4, fold=-1)
from qiskit.providers.fake_provider import GenericBackendV2
backend = GenericBackendV2(
2 * norb, basis_gates=["cp", "xx_plus_yy", "p", "x"]
)
Étape 2 : Optimiser le problème pour l'exécution quantique
Ensuite, nous optimisons le circuit pour un matériel cible. Pour l'instant, nous allons créer un backend générique avec un nombre de qubits spécifié et un ensemble de portes vers lesquelles les circuits d'évolution temporelle se décomposent naturellement.
from qiskit.transpiler import generate_preset_pass_manager
pass_manager = generate_preset_pass_manager(
optimization_level=3, backend=backend
)
isa_circuits = pass_manager.run(circuits)
from qiskit.visualization import plot_histogram
from qiskit.primitives import StatevectorSampler
# Sample from the circuits
sampler = StatevectorSampler()
job = sampler.run(isa_circuits, shots=500)
Maintenant, nous utilisons Qiskit pour transpiler les circuits vers le backend cible.
from qiskit.primitives import BitArray
# Combine the shots from the individual Trotter circuits
bit_array = BitArray.concatenate_shots(
[result.data.meas for result in job.result()]
)
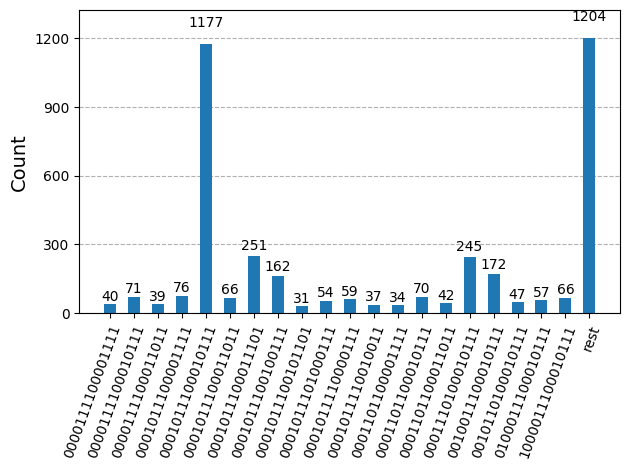
plot_histogram(bit_array.get_counts(), number_to_keep=20)
Étape 3 : Exécuter avec les primitives Qiskit
Après avoir optimisé les circuits pour l'exécution matérielle, nous sommes prêts à les exécuter sur le matériel cible et à collecter des échantillons pour l'estimation de l'énergie de l'état fondamental. Après avoir utilisé la primitive Sampler pour échantillonner des chaînes de bits à partir de chaque circuit, nous combinons tous les résultats dans un seul dictionnaire de comptages et traçons les 20 chaînes de bits les plus fréquemment échantillonnées.
from qiskit_addon_sqd.fermion import (
SCIResult,
diagonalize_fermionic_hamiltonian,
)
# List to capture intermediate results
result_history = []
def callback(results: list[SCIResult]):
result_history.append(results)
iteration = len(result_history)
print(f"Iteration {iteration}")
for i, result in enumerate(results):
print(f"\tSubsample {i}")
print(f"\t\tEnergy: {result.energy}")
print(
f"\t\tSubspace dimension: {np.prod(result.sci_state.amplitudes.shape)}"
)
rng = np.random.default_rng(24)
result = diagonalize_fermionic_hamiltonian(
h1e_momentum,
h2e_momentum,
bit_array,
samples_per_batch=100,
norb=norb,
nelec=nelec,
num_batches=3,
max_iterations=5,
symmetrize_spin=True,
callback=callback,
seed=rng,
)
Iteration 1
Subsample 0
Energy: -13.4222953188441
Subspace dimension: 529
Subsample 1
Energy: -13.42237556285828
Subspace dimension: 784
Subsample 2
Energy: -13.422045397387413
Subspace dimension: 529
Iteration 2
Subsample 0
Energy: -13.422379583305478
Subspace dimension: 900
Subsample 1
Energy: -13.422376197704326
Subspace dimension: 841
Subsample 2
Energy: -13.422421162849295
Subspace dimension: 1089
Iteration 3
Subsample 0
Energy: -13.422421164670345
Subspace dimension: 1156
Subsample 1
Energy: -13.422421492737689
Subspace dimension: 1156
Subsample 2
Energy: -13.422421205869572
Subspace dimension: 1156
Iteration 4
Subsample 0
Energy: -13.422421494558726
Subspace dimension: 1225
Subsample 1
Energy: -13.422421492737689
Subspace dimension: 1156
Subsample 2
Energy: -13.422421492737689
Subspace dimension: 1156
Étape 4 : Post-traiter et renvoyer le résultat au format classique souhaité
Nous exécutons maintenant l'algorithme SQD à l'aide de la fonction diagonalize_fermionic_hamiltonian. Consulte la documentation de l'API pour des explications sur les arguments de cette fonction.
import matplotlib.pyplot as plt
min_es = [
min(result, key=lambda res: res.energy).energy
for result in result_history
]
min_id, min_e = min(enumerate(min_es), key=lambda x: x[1])
# Data for energies plot
x1 = range(len(result_history))
# Data for avg spatial orbital occupancy
y2 = np.sum(result.orbital_occupancies, axis=0)
x2 = range(len(y2))
fig, axs = plt.subplots(1, 2, figsize=(12, 6))
# Plot energies
axs[0].plot(x1, min_es, label="energy", marker="o")
axs[0].set_xticks(x1)
axs[0].set_xticklabels(x1)
axs[0].axhline(
y=reference_energy,
color="#BF5700",
linestyle="--",
label="reference energy",
)
axs[0].set_title("Approximated Ground State Energy vs SQD Iterations")
axs[0].set_xlabel("Iteration Index", fontdict={"fontsize": 12})
axs[0].set_ylabel("Energy", fontdict={"fontsize": 12})
axs[0].legend()
# Plot orbital occupancy
axs[1].bar(x2, y2, width=0.8)
axs[1].set_xticks(x2)
axs[1].set_xticklabels(x2)
axs[1].set_title("Avg Occupancy per Spatial Orbital")
axs[1].set_xlabel("Orbital Index", fontdict={"fontsize": 12})
axs[1].set_ylabel("Avg Occupancy", fontdict={"fontsize": 12})
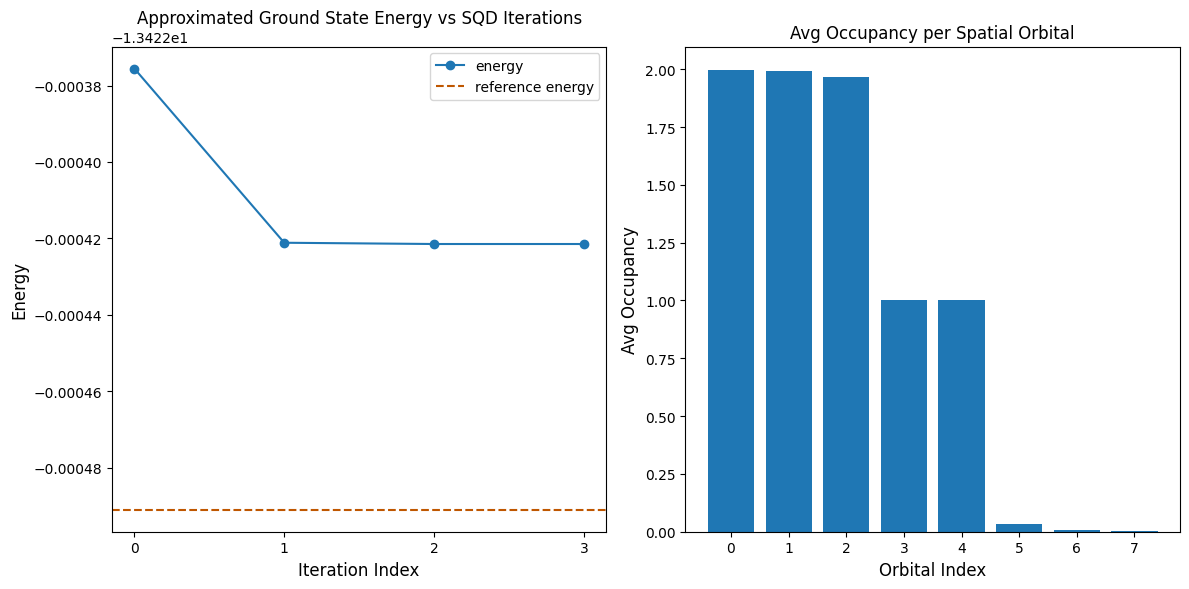
print(f"Reference energy: {reference_energy:.5f}")
print(f"SQD energy: {min_e:.5f}")
print(f"Absolute error: {abs(min_e - reference_energy):.5f}")
plt.tight_layout()
plt.show()
Reference energy: -13.42249
SQD energy: -13.42242
Absolute error: 0.00007
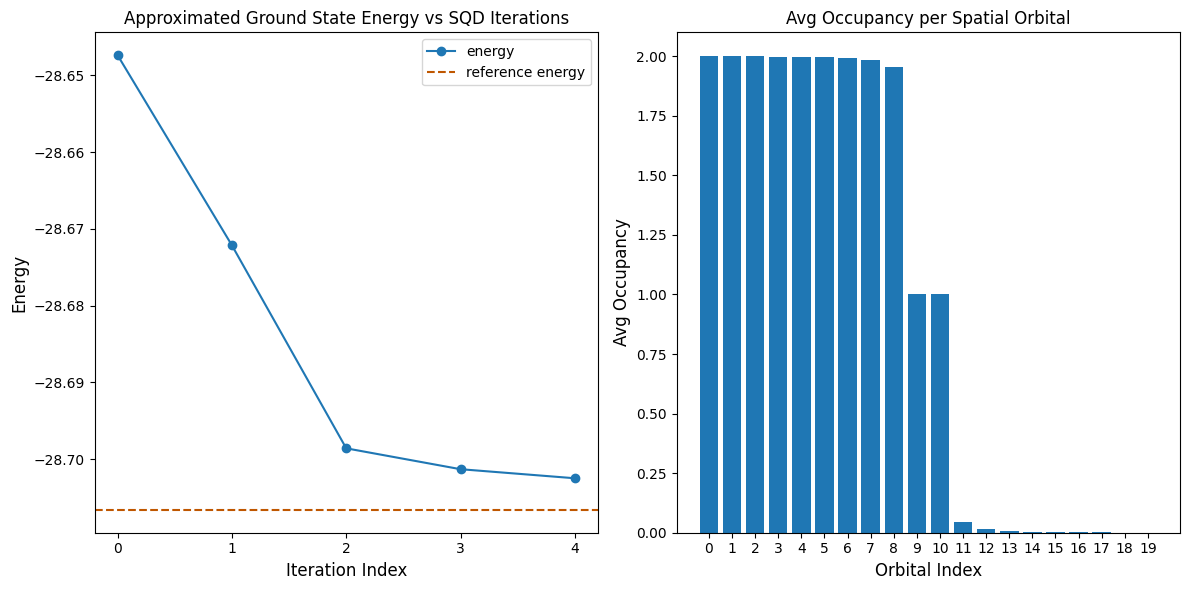
La cellule de code suivante trace les résultats. Le premier graphique montre l'énergie calculée en fonction du nombre d'itérations de récupération de configuration, et le second graphique montre l'occupation moyenne de chaque orbitale spatiale après la dernière itération. Comme il s'agit d'un si petit problème, la première itération nous amène déjà très près de l'énergie exacte (note l'échelle de l'axe y).
rdm1 = result.sci_state.rdm(rank=1, spin_summed=True)
rdm2 = result.sci_state.rdm(rank=2, spin_summed=True)
energy = np.sum(h1e_momentum * rdm1) + 0.5 * np.sum(h2e_momentum * rdm2)
print(f"Recomputed energy: {energy:.5f}")
Recomputed energy: -13.42242
Vérifier l'énergie
L'énergie renvoyée par la SQD est garantie d'être une borne supérieure de la véritable énergie de l'état fondamental. La valeur de l'énergie peut être vérifiée car la SQD renvoie également les coefficients du vecteur d'état approximant l'état fondamental. Tu peux calculer l'énergie à partir du vecteur d'état en utilisant ses matrices de densité réduites à une et deux particules, comme le montre la cellule de code suivante.
Exemple à grande échelle sur matériel réel
Nous exécutons maintenant un exemple plus grand sur un vrai QPU. Pour l'énergie de référence, nous utilisons les résultats d'un calcul DMRG effectué séparément.
from qiskit_ibm_runtime import SamplerV2 as Sampler
from qiskit_ibm_runtime import QiskitRuntimeService
# Model parameters
norb = 20
nelec = (norb // 2, norb // 2)
n_bath = norb - 1
hybridization = 1.0
hopping = 1.0
onsite = 10.0
chemical_potential = -0.5 * onsite
# Generate Hamiltonian and orbital rotation
h1e, h2e = siam_hamiltonian(
norb=norb,
hopping=hopping,
onsite=onsite,
hybridization=hybridization,
chemical_potential=chemical_potential,
)
orbital_rotation = momentum_basis(norb)
h1e_momentum, h2e_momentum = rotated(h1e, h2e, orbital_rotation.T.conj())
impurity_index = n_bath // 2
# Set reference energy to DMRG value computed separately
reference_energy = -28.70659686
# Algorithm parameters
time_step = 0.2
krylov_dim = 8
# Construct circuits
qubits = QuantumRegister(2 * norb, name="q")
circuit = QuantumCircuit(qubits)
for instruction in prepare_initial_state(qubits, norb=norb, nocc=norb // 2):
circuit.append(instruction)
circuit.measure_all()
circuits = [circuit.copy()]
one_body_evolution = scipy.linalg.expm(-1j * time_step * h1e_momentum)
for i in range(krylov_dim - 1):
circuit.remove_final_measurements()
for instruction in trotter_step(
qubits,
time_step,
one_body_evolution,
h2e_momentum,
impurity_index,
norb,
):
circuit.append(instruction)
circuit.measure_all()
circuits.append(circuit.copy())
# Initialize hardware backend
service = QiskitRuntimeService()
backend = service.least_busy(
operational=True, simulator=False, min_num_qubits=127
)
print(f"Using backend {backend.name}")
# Transpile to backend
pass_manager = generate_preset_pass_manager(
optimization_level=3, backend=backend
)
isa_circuits = pass_manager.run(circuits)
# Sample from the circuits
sampler = Sampler(backend)
sampler.options.environment.job_tags = ["TUT_SKQD"]
job = sampler.run(isa_circuits, shots=500)
# Combine the shots from the individual Trotter circuits
bit_array = BitArray.concatenate_shots(
[result.data.meas for result in job.result()]
)
# Run configuration recovery and diagonalization
result_history = []
def callback(results: list[SCIResult]):
result_history.append(results)
iteration = len(result_history)
print(f"Iteration {iteration}")
for i, result in enumerate(results):
print(f"\tSubsample {i}")
print(f"\t\tEnergy: {result.energy}")
print(
f"\t\tSubspace dimension: {np.prod(result.sci_state.amplitudes.shape)}"
)
rng = np.random.default_rng(24)
result = diagonalize_fermionic_hamiltonian(
h1e_momentum,
h2e_momentum,
bit_array,
samples_per_batch=100,
norb=norb,
nelec=nelec,
num_batches=3,
max_iterations=5,
symmetrize_spin=True,
callback=callback,
seed=rng,
)
# Plot results
min_es = [
min(result, key=lambda res: res.energy).energy
for result in result_history
]
min_id, min_e = min(enumerate(min_es), key=lambda x: x[1])
x1 = range(len(result_history))
y2 = np.sum(result.orbital_occupancies, axis=0)
x2 = range(len(y2))
fig, axs = plt.subplots(1, 2, figsize=(12, 6))
axs[0].plot(x1, min_es, label="energy", marker="o")
axs[0].set_xticks(x1)
axs[0].set_xticklabels(x1)
axs[0].axhline(
y=reference_energy,
color="#BF5700",
linestyle="--",
label="reference energy",
)
axs[0].set_title("Approximated Ground State Energy vs SQD Iterations")
axs[0].set_xlabel("Iteration Index", fontdict={"fontsize": 12})
axs[0].set_ylabel("Energy", fontdict={"fontsize": 12})
axs[0].legend()
axs[1].bar(x2, y2, width=0.8)
axs[1].set_xticks(x2)
axs[1].set_xticklabels(x2)
axs[1].set_title("Avg Occupancy per Spatial Orbital")
axs[1].set_xlabel("Orbital Index", fontdict={"fontsize": 12})
axs[1].set_ylabel("Avg Occupancy", fontdict={"fontsize": 12})
print(f"Reference energy: {reference_energy:.5f}")
print(f"SQD energy: {min_e:.5f}")
print(f"Absolute error: {abs(min_e - reference_energy):.5f}")
plt.tight_layout()
plt.show()
Using backend ibm_boston
Iteration 1
Subsample 0
Energy: -28.63965951544449
Subspace dimension: 9801
Subsample 1
Energy: -28.625588929202006
Subspace dimension: 9409
Subsample 2
Energy: -28.647371834135498
Subspace dimension: 8281
Iteration 2
Subsample 0
Energy: -28.67213260849567
Subspace dimension: 29584
Subsample 1
Energy: -28.670340686158816
Subspace dimension: 27225
Subsample 2
Energy: -28.669976379525988
Subspace dimension: 31329
Iteration 3
Subsample 0
Energy: -28.68622875601382
Subspace dimension: 36100
Subsample 1
Energy: -28.698569623143126
Subspace dimension: 34225
Subsample 2
Energy: -28.694848533971882
Subspace dimension: 33856
Iteration 4
Subsample 0
Energy: -28.69883392844593
Subspace dimension: 42025
Subsample 1
Energy: -28.701289495200996
Subspace dimension: 38025
Subsample 2
Energy: -28.699319594978245
Subspace dimension: 45369
Iteration 5
Subsample 0
Energy: -28.701936886834154
Subspace dimension: 51076
Subsample 1
Energy: -28.702468711812013
Subspace dimension: 53824
Subsample 2
Energy: -28.702298147575938
Subspace dimension: 52900
Reference energy: -28.70660
SQD energy: -28.70247
Absolute error: 0.00413
Prochaines étapes
Si tu as trouvé ce travail intéressant, tu pourrais être intéressé par les ressources suivantes :
- Diagonalisation quantique basée sur les échantillons d'un hamiltonien de chimie - un tutoriel connexe utilisant un ansatz variationnel heuristique plutôt que des circuits de Trotter
- Diagonalisation quantique de Krylov des hamiltoniens de réseau - un tutoriel sur la méthode KQD
- Documentation de l'API de l'addon SQD - référence pour la fonction
diagonalize_fermionic_hamiltonian - Quantum-Centric Algorithm for Sample-Based Krylov Diagonalization - l'article sur lequel ce tutoriel est basé