Diagonalisation quantique basée sur des échantillons d'un Hamiltonien de chimie
Estimation d'utilisation : moins d'une minute sur un processeur Heron r2 (REMARQUE : Il s'agit d'une estimation uniquement. Ton temps d'exécution peut varier.)
Objectifs d'apprentissage
Après ce tutoriel, les utilisateurs devraient comprendre :
- Comment utiliser l'extension SQD de Qiskit pour approximer l'énergie de l'état fondamental d'un système moléculaire à partir de chaînes de bits échantillonnées sur une unité de traitement quantique (QPU).
- Comment utiliser ffsim pour construire un circuit ansatz local unitary cluster Jastrow (LUCJ) pour la simulation de chimie quantique.
Prérequis
Nous recommandons aux utilisateurs de se familiariser avec les sujets suivants avant de suivre ce tutoriel :
- Chimie quantique et seconde quantification
- Utilisation de la primitive Sampler pour échantillonner des circuits quantiques
Contexte
Dans ce tutoriel, nous montrons comment post-traiter des échantillons quantiques bruités pour approximer l'état fondamental de la molécule d'azote à la longueur de liaison d'équilibre, en utilisant l'extension SQD de Qiskit pour implémenter l'algorithme de diagonalisation quantique basée sur des échantillons (SQD). Plus de détails sur le logiciel sont disponibles dans la documentation correspondante, notamment un exemple simple pour démarrer.
Ce tutoriel est recommandé pour les utilisateurs familiers avec la chimie quantique : en particulier, la familiarité avec la recherche des énergies d'état fondamental d'une molécule. Pour un guide détaillé du workflow, consulte le cours sur les algorithmes de diagonalisation quantique.
SQD est une technique permettant de trouver les valeurs propres et les vecteurs propres d'opérateurs quantiques, tels qu'un Hamiltonien de système quantique, en combinant calcul quantique et calcul classique distribué. Le calcul classique distribué est utilisé pour traiter les échantillons obtenus à partir d'un processeur quantique, et pour projeter et diagonaliser un Hamiltonien cible dans un sous-espace qu'ils engendrent. Un workflow basé sur SQD comporte les étapes suivantes :
- Choisir un ansatz de circuit et l'appliquer sur un ordinateur quantique à un état de référence (dans ce cas, l'état de Hartree-Fock).
- Échantillonner des chaînes de bits à partir de l'état quantique résultant.
- Exécuter la procédure de récupération auto-cohérente des configurations sur les chaînes de bits pour obtenir l'approximation de l'état fondamental.
SQD est reconnu pour bien fonctionner lorsque l'état propre cible est creux : la fonction d'onde est supportée dans un ensemble d'états de base dont la taille n'augmente pas exponentiellement avec la taille du problème.
Chimie quantique
Le Hamiltonien d'un système moléculaire peut s'écrire sous la forme
où les et sont des nombres complexes appelés intégrales moléculaires qui peuvent être calculés à partir de la spécification de la molécule à l'aide d'un programme informatique. Dans ce tutoriel, nous calculons les intégrales à l'aide du logiciel PySCF.
Pour plus de détails sur la dérivation du Hamiltonien moléculaire, consulte un manuel de chimie quantique (par exemple, Modern Quantum Chemistry de Szabo et Ostlund). Pour une explication de haut niveau sur la façon dont les problèmes de chimie quantique sont transposés sur des ordinateurs quantiques, consulte le cours Mapping Problems to Qubits de la Qiskit Global Summer School 2024.
Ansatz local unitary cluster Jastrow (LUCJ)
SQD nécessite un ansatz de circuit quantique pour en tirer des échantillons. Dans ce tutoriel, nous utiliserons l'ansatz local unitary cluster Jastrow (LUCJ) en raison de sa combinaison de motivation physique et d'adéquation au matériel. Nous utiliserons ffsim pour construire le circuit ansatz.
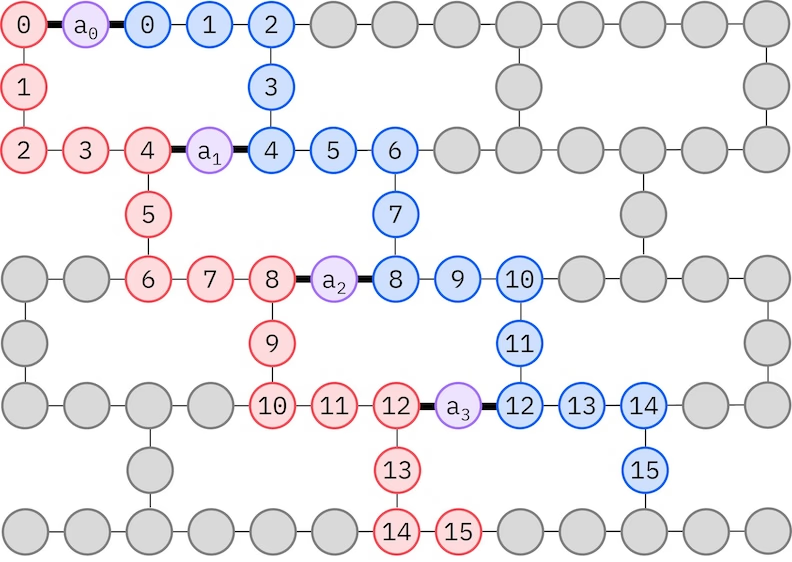
L'ansatz LUCJ s'adapte aux QPUs avec une connectivité limitée entre les qubits. Les orbitales de spin sont associées à des qubits de sorte que l'ansatz ne nécessite pas de routage avec des portes SWAP. Le matériel IBM® possède une topologie de qubits en réseau heavy-hex, auquel cas nous pouvons adopter un motif en « zigzag », illustré ci-dessous. Dans ce motif, les orbitales de même spin sont associées à des qubits ayant une topologie linéaire (cercles rouges et bleus), et une connexion entre les orbitales de spin différent est présente tous les 4 orbitales spatiales, la connexion étant facilitée par un qubit ancillaire (cercles violets).
Récupération auto-cohérente des configurations
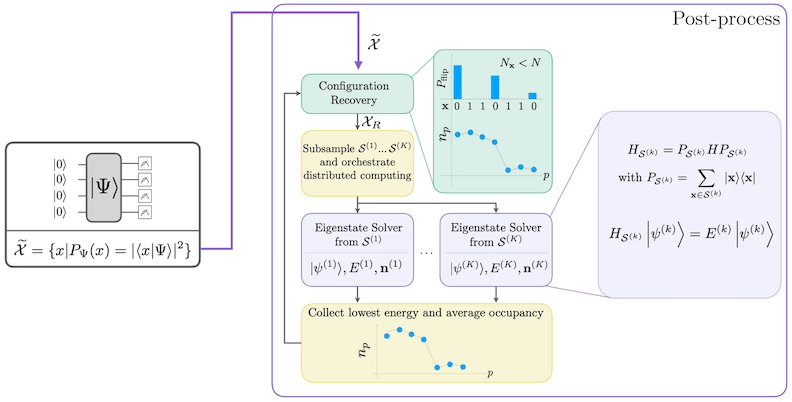
La procédure de récupération auto-cohérente des configurations est conçue pour extraire autant de signal que possible des échantillons quantiques bruités. Étant donné que le Hamiltonien moléculaire conserve le nombre de particules et le spin Z, il est logique de choisir un ansatz de circuit qui conserve également ces symétries. Lorsqu'il est appliqué à l'état de Hartree-Fock, l'état résultant a un nombre de particules et un spin Z fixes dans le cas sans bruit. Par conséquent, les moitiés spin- et spin- de toute chaîne de bits échantillonnée à partir de cet état devraient avoir le même poids de Hamming que dans l'état de Hartree-Fock. En raison de la présence de bruit dans les processeurs quantiques actuels, certaines chaînes de bits mesurées violeront cette propriété. Une forme simple de post-sélection consisterait à écarter ces chaînes de bits, mais cela serait un gaspillage car elles pourraient encore contenir du signal. La procédure de récupération auto-cohérente tente de récupérer une partie de ce signal en post-traitement. La procédure est itérative et nécessite en entrée une estimation des occupations moyennes de chaque orbitale dans l'état fondamental, qui est d'abord calculée à partir des échantillons bruts. La procédure est exécutée en boucle, et chaque itération comporte les étapes suivantes :
- Pour chaque chaîne de bits qui viole les symétries spécifiées, inverser ses bits selon une procédure probabiliste conçue pour rapprocher la chaîne de bits de l'estimation actuelle des occupations orbitales moyennes, afin d'obtenir une nouvelle chaîne de bits.
- Rassembler toutes les anciennes et nouvelles chaînes de bits satisfaisant les symétries, et sous-échantillonner des sous-ensembles d'une taille fixe, choisie à l'avance.
- Pour chaque sous-ensemble de chaînes de bits, projeter le Hamiltonien dans le sous-espace engendré par les vecteurs de base correspondants (voir la section précédente pour une description de ces vecteurs de base), et calculer une estimation de l'état fondamental du Hamiltonien projeté sur un ordinateur classique.
- Mettre à jour l'estimation des occupations orbitales moyennes avec l'estimation de l'état fondamental ayant l'énergie la plus basse.
Diagramme du workflow SQD
Le workflow SQD est représenté dans le diagramme suivant :
Configuration requise
Avant de commencer ce tutoriel, assure-toi d'avoir installé les éléments suivants :
- Qiskit SDK v1.0 ou ultérieur, avec le support de visualisation
- Qiskit Runtime v0.22 ou ultérieur (
pip install qiskit-ibm-runtime) - Extension SQD de Qiskit v0.11 ou ultérieur (
pip install qiskit-addon-sqd) - ffsim v0.0.75 ou ultérieur (
pip install ffsim)
Configuration
# Added by doQumentation — required packages for this notebook
!pip install -q ffsim matplotlib numpy pyscf qiskit qiskit-addon-sqd qiskit-ibm-runtime
import math
import ffsim
import matplotlib.pyplot as plt
import numpy as np
import pyscf
import pyscf.cc
import pyscf.mcscf
from qiskit import QuantumCircuit, QuantumRegister
from qiskit.primitives import StatevectorSampler
from qiskit.providers.fake_provider import GenericBackendV2
from qiskit_ibm_runtime import QiskitRuntimeService
from qiskit_ibm_runtime import SamplerV2 as Sampler
Exemple à petite échelle avec simulateur
Dans ce tutoriel, nous trouverons une approximation de l'état fondamental de la molécule d'azote à sa longueur de liaison d'équilibre. Nous utilisons d'abord un petit ensemble de base STO-6G afin de pouvoir simuler l'expérience et vérifier qu'elle fonctionne correctement.
Étape 1 : Transposer les entrées classiques en un problème quantique
Tout d'abord, nous spécifions la molécule et ses propriétés.
# Specify molecule properties
spin_sq = 0
# Build N2 molecule
mol = pyscf.gto.Mole()
mol.build(
atom=[["N", (0, 0, 0)], ["N", (1.0, 0, 0)]],
basis="sto-6g",
symmetry="Dooh",
)
# Define active space
n_frozen = 2
active_space = range(n_frozen, mol.nao_nr())
# Get molecular integrals
scf = pyscf.scf.RHF(mol).run()
norb = len(active_space)
n_electrons = int(sum(scf.mo_occ[active_space]))
n_alpha = (n_electrons + mol.spin) // 2
n_beta = (n_electrons - mol.spin) // 2
nelec = (n_alpha, n_beta)
cas = pyscf.mcscf.CASCI(scf, norb, nelec)
mo = cas.sort_mo(active_space, base=0)
hcore, nuclear_repulsion_energy = cas.get_h1cas(mo)
eri = pyscf.ao2mo.restore(1, cas.get_h2cas(mo), norb)
# Compute exact energy using FCI
reference_energy = cas.run().e_tot
print(f"norb = {norb}")
print(f"nelec = {nelec}")
converged SCF energy = -108.464957764796
CASCI E = -108.595987350986 E(CI) = -32.4115475088426 S^2 = 0.0000000
norb = 8
nelec = (5, 5)
Avant de construire le circuit d'ansatz LUCJ, nous effectuons d'abord un calcul CCSD dans la cellule de code suivante. Les amplitudes et issues de ce calcul seront utilisées pour initialiser les paramètres de l'ansatz.
# Get CCSD t2 amplitudes for initializing the ansatz
ccsd = pyscf.cc.CCSD(
scf, frozen=[i for i in range(mol.nao_nr()) if i not in active_space]
).run()
t1 = ccsd.t1
t2 = ccsd.t2
E(CCSD) = -108.5933309085008 E_corr = -0.1283731437052354
Maintenant, nous utilisons ffsim pour créer le circuit ansatz. Comme notre molécule a un état de Hartree-Fock à couche fermée, nous utilisons la variante équilibrée en spin de l'ansatz UCJ, UCJOpSpinBalanced. Nous définissons optimize=True dans la méthode from_t_amplitudes pour activer la double-factorisation « compressée » des amplitudes (voir The local unitary cluster Jastrow (LUCJ) ansatz dans la documentation de ffsim pour plus de détails).
Comme l'ansatz LUCJ s'adapte à la connectivité disponible du QPU, nous devons initialiser le backend QPU avant de créer l'ansatz. Pour l'instant, nous créons un backend générique avec une carte de couplage heavy-hex et un ensemble de portes auquel l'ansatz LUCJ se décompose naturellement. Ensuite, nous utilisons ffsim.qiskit.generate_lucj_pass_manager pour créer un gestionnaire de passes spécialisé dans la transpilation de l'ansatz LUCJ vers le backend donné selon le schéma en « zigzag » décrit dans la section de contexte sur l'ansatz LUCJ. Cette fonction utilise une heuristique de scoring pour minimiser les erreurs associées au schéma sélectionné, ce qui est important si ton backend est un vrai QPU, ou un simulateur avec un modèle de bruit. En plus de retourner le gestionnaire de passes, cette fonction retourne également les paires de couplage alpha-bêta qui peuvent être implémentées sur le matériel. Si toutes les paires ne peuvent pas être implémentées, elle émet un avertissement.
import warnings
from qiskit.transpiler import CouplingMap
warnings.formatwarning = lambda msg, *args, **kwargs: f"Warning: {msg}\n"
# Set ansatz properties
n_reps = 1
pairs_aa = [(p, p + 1) for p in range(norb - 1)]
# Let generate_lucj_pass_manager determine the alpha-beta interactions
pairs_ab = None
# Initialize backend
coupling_map = CouplingMap.from_heavy_hex(3)
backend = GenericBackendV2(
coupling_map.size(),
coupling_map=coupling_map,
basis_gates=["cp", "xx_plus_yy", "p", "x", "swap"],
)
# Create pass manager
pass_manager, pairs_ab = ffsim.qiskit.generate_lucj_pass_manager(
backend=backend,
norb=norb,
connectivity="heavy-hex",
interaction_pairs=(pairs_aa, pairs_ab),
optimization_level=3,
)
# Create the LUCJ ansatz operator
ucj_op = ffsim.UCJOpSpinBalanced.from_t_amplitudes(
t2=t2,
t1=t1,
n_reps=n_reps,
interaction_pairs=(pairs_aa, pairs_ab),
# Setting optimize=True enables the "compressed" factorization
optimize=True,
# Limit the number of optimization iterations to prevent the code cell
# from running too long. Removing this line may improve results.
options=dict(maxiter=1000),
)
# create an empty quantum circuit
qubits = QuantumRegister(2 * norb, name="q")
circuit = QuantumCircuit(qubits)
# prepare Hartree-Fock state as the reference state and append it
# to the quantum circuit
circuit.append(ffsim.qiskit.PrepareHartreeFockJW(norb, nelec), qubits)
# apply the UCJ operator to the reference state
circuit.append(ffsim.qiskit.UCJOpSpinBalancedJW(ucj_op), qubits)
circuit.measure_all()
Étape 2 : Optimiser le circuit pour l'exécution sur du matériel quantique
Ensuite, nous optimisons le circuit pour un matériel cible. En général, cette étape implique d'initialiser le backend matériel et un gestionnaire de passes pour ce backend. Cependant, comme l'ansatz LUCJ est adapté à la connectivité du matériel, nous avons déjà effectué ces actions à l'étape précédente. Il ne reste plus qu'à exécuter le gestionnaire de passes sur le circuit pour le transpiler en un circuit ISA qui peut être directement exécuté sur le QPU.
isa_circuit = pass_manager.run(circuit)
print(f"Gate counts: {isa_circuit.count_ops()}")
Gate counts: OrderedDict({'xx_plus_yy': 86, 'p': 16, 'measure': 16, 'cp': 15, 'x': 10, 'swap': 2, 'barrier': 1})
Étape 3 : Exécuter à l'aide des primitives Qiskit
Après avoir optimisé le circuit pour l'exécution sur matériel, nous sommes prêts à l'exécuter sur le matériel cible et à collecter des échantillons pour l'estimation de l'énergie de l'état fondamental. Comme nous n'avons qu'un seul circuit, nous utilisons le mode d'exécution par Job de Qiskit Runtime et exécutons notre circuit.
rng = np.random.default_rng()
sampler = StatevectorSampler(seed=rng)
job = sampler.run([isa_circuit], shots=100_000)
Warning: Trying to add QuantumRegister to a QuantumCircuit having a layout
primitive_result = job.result()
pub_result = primitive_result[0]
Étape 4 : Post-traiter et renvoyer le résultat dans le format classique souhaité
Une métrique utile pour évaluer la qualité de la sortie du QPU est le nombre de configurations valides retournées. Une configuration valide a le nombre de particules et le spin Z corrects, ce qui signifie que la moitié droite de la chaîne de bits a un poids de Hamming égal au nombre d'électrons de spin-up, et la moitié gauche a un poids de Hamming égal au nombre d'électrons de spin-down. La cellule suivante calcule la fraction des configurations échantillonnées qui sont valides.
def is_valid_bitstring(
bitstring: str, norb: int, nelec: tuple[int, int]
) -> bool:
n_alpha, n_beta = nelec
return (
len(bitstring) == 2 * norb
and bitstring[norb:].count("1") == n_alpha
and bitstring[:norb].count("1") == n_beta
)
bit_array = pub_result.data.meas
num_valid = sum(
is_valid_bitstring(b, norb, nelec) for b in bit_array.get_bitstrings()
)
valid_fraction = num_valid / bit_array.num_shots
print(f"Fraction of sampled configurations that are valid: {valid_fraction}")
Fraction of sampled configurations that are valid: 1.0
Toutes les chaînes de bits sont valides car nous échantillonnons le circuit sur un simulateur sans bruit. Lors de l'exécution sur un QPU bruité, la fraction sera inférieure à un, mais on espère qu'elle sera plus grande que la fraction attendue si les chaînes de bits étaient échantillonnées de manière uniformément aléatoire, ce qui est calculé dans la cellule suivante.
expected_fraction_random = (
math.comb(norb, n_alpha) * math.comb(norb, n_beta) / 2 ** (2 * norb)
)
print(
f"Expected fraction of valid configurations from uniformly random bitstrings: "
f"{expected_fraction_random}"
)
Expected fraction of valid configurations from uniformly random bitstrings: 0.0478515625
Maintenant, nous estimons l'énergie de l'état fondamental du Hamiltonien à l'aide de la fonction diagonalize_fermionic_hamiltonian. Cette fonction effectue la procédure de récupération auto-cohérente des configurations pour affiner itérativement les échantillons quantiques bruités et améliorer l'estimation de l'énergie. Nous passons une fonction de rappel afin de pouvoir sauvegarder les résultats intermédiaires pour une analyse ultérieure. Voir la documentation API pour les explications des arguments de diagonalize_fermionic_hamiltonian.
Ici, nous utilisons l'argument initial_occupancies de diagonalize_fermionic_hamiltonian pour spécifier la configuration de Hartree-Fock comme estimation initiale des occupations orbitales dans l'état fondamental. Cette approche est sensée pour les systèmes où l'état fondamental a un support significatif sur la configuration de Hartree-Fock, mais elle peut ne pas être appropriée dans d'autres situations, bien que des méthodes de calcul plus avancées puissent produire de meilleures estimations initiales dans ces cas. La spécification de initial_occupancies permet également à la récupération des configurations de s'exécuter même si aucune configuration valide n'a été échantillonnée, comme cela peut être le cas lors de l'échantillonnage d'un grand circuit sur un QPU bruité. Sans cet argument, la récupération des configurations échouerait et lèverait une erreur si aucune configuration valide n'était fournie.
from functools import partial
from qiskit_addon_sqd.fermion import (
SCIResult,
diagonalize_fermionic_hamiltonian,
solve_sci_batch,
)
# SQD options
energy_tol = 1e-3
occupancies_tol = 1e-3
max_iterations = 5
# Eigenstate solver options
num_batches = 3
samples_per_batch = 300
symmetrize_spin = True
carryover_threshold = 1e-4
max_cycle = 200
# Use the Hartree-Fock configuration as an initial guess for the orbital occupancies
initial_occupancies = (
np.array([1] * n_alpha + [0] * (norb - n_alpha)),
np.array([1] * n_beta + [0] * (norb - n_beta)),
)
# Pass options to the built-in eigensolver. If you just want to use the defaults,
# you can omit this step, in which case you would not specify the sci_solver argument
# in the call to diagonalize_fermionic_hamiltonian below.
sci_solver = partial(solve_sci_batch, spin_sq=0.0, max_cycle=max_cycle)
# List to capture intermediate results
result_history = []
def callback(results: list[SCIResult]):
result_history.append(results)
iteration = len(result_history)
print(f"Iteration {iteration}")
for i, result in enumerate(results):
print(f"\tSubsample {i}")
print(f"\t\tEnergy: {result.energy + nuclear_repulsion_energy}")
print(
f"\t\tSubspace dimension: {np.prod(result.sci_state.amplitudes.shape)}"
)
result = diagonalize_fermionic_hamiltonian(
hcore,
eri,
bit_array,
samples_per_batch=samples_per_batch,
norb=norb,
nelec=nelec,
num_batches=num_batches,
energy_tol=energy_tol,
occupancies_tol=occupancies_tol,
max_iterations=max_iterations,
sci_solver=sci_solver,
symmetrize_spin=symmetrize_spin,
initial_occupancies=initial_occupancies,
carryover_threshold=carryover_threshold,
callback=callback,
seed=rng,
)
final_energy = result.energy + nuclear_repulsion_energy
energy_error = final_energy - reference_energy
print(f"Final energy: {final_energy}")
print(f"Final energy error: {energy_error}")
Iteration 1
Subsample 0
Energy: -108.59275573641656
Subspace dimension: 900
Subsample 1
Energy: -108.59275573641656
Subspace dimension: 900
Subsample 2
Energy: -108.59275573641656
Subspace dimension: 900
Iteration 2
Subsample 0
Energy: -108.59275573641656
Subspace dimension: 900
Subsample 1
Energy: -108.59275573641656
Subspace dimension: 900
Subsample 2
Energy: -108.59275573641656
Subspace dimension: 900
Final energy: -108.59275573641656
Final energy error: 0.0032316145694579745
Visualiser les résultats
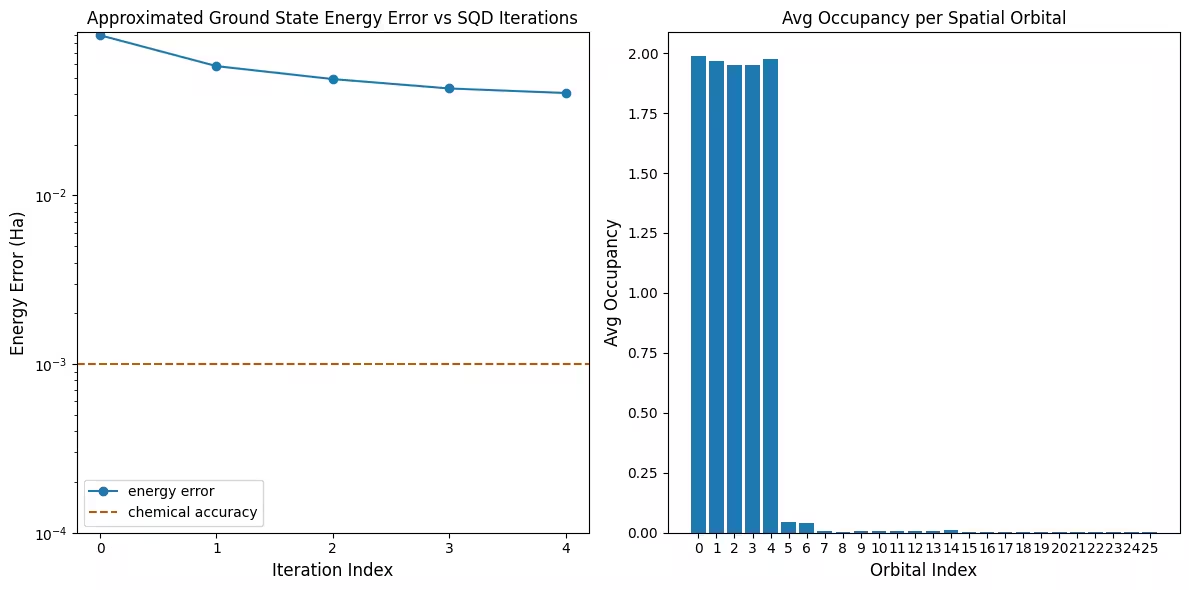
Le premier graphique montre que dans cette simulation, nous sommes déjà à moins de 1 mH de la réponse exacte après la première itération (la précision chimique est généralement acceptée à 1 kcal/mol 1.6 mH). C'est un petit système, cependant, et comme les échantillons sont sans bruit, la récupération des configurations n'est pas nécessaire. Sur un système plus grand exécuté sur un QPU bruité, plusieurs itérations de récupération des configurations peuvent être nécessaires, et la précision finale peut être moins bonne. En général, l'énergie peut être améliorée en autorisant plus d'itérations de récupération des configurations ou en augmentant le nombre d'échantillons par lot.
Le second graphique montre l'occupation moyenne de chaque orbitale spatiale après l'itération finale. Nous pouvons voir que les électrons de spin-up et de spin-down occupent les cinq premières orbitales avec une probabilité élevée dans nos solutions.
# Data for energies plot
x1 = range(len(result_history))
min_e = [
min(result, key=lambda res: res.energy).energy + nuclear_repulsion_energy
for result in result_history
]
e_diff = [abs(e - reference_energy) for e in min_e]
yt1 = [1.0, 1e-1, 1e-2, 1e-3, 1e-4]
# Chemical accuracy (+/- 1 milli-Hartree)
chem_accuracy = 0.001
# Data for avg spatial orbital occupancy
y2 = np.sum(result.orbital_occupancies, axis=0)
x2 = range(len(y2))
fig, axs = plt.subplots(1, 2, figsize=(12, 6))
# Plot energies
axs[0].plot(x1, e_diff, label="energy error", marker="o")
axs[0].set_xticks(x1)
axs[0].set_xticklabels(x1)
axs[0].set_yticks(yt1)
axs[0].set_yticklabels(yt1)
axs[0].set_yscale("log")
axs[0].set_ylim(1e-4)
axs[0].axhline(
y=chem_accuracy,
color="#BF5700",
linestyle="--",
label="chemical accuracy",
)
axs[0].set_title("Approximated Ground State Energy Error vs SQD Iterations")
axs[0].set_xlabel("Iteration Index", fontdict={"fontsize": 12})
axs[0].set_ylabel("Energy Error (Ha)", fontdict={"fontsize": 12})
axs[0].legend()
# Plot orbital occupancy
axs[1].bar(x2, y2, width=0.8)
axs[1].set_xticks(x2)
axs[1].set_xticklabels(x2)
axs[1].set_title("Avg Occupancy per Spatial Orbital")
axs[1].set_xlabel("Orbital Index", fontdict={"fontsize": 12})
axs[1].set_ylabel("Avg Occupancy", fontdict={"fontsize": 12})
plt.tight_layout()
plt.show()

Exemple à grande échelle sur du matériel réel
Maintenant, nous exécutons un exemple plus grand sur du vrai matériel quantique. Ici, nous dérivons un espace actif pour la molécule d'azote à partir de l'ensemble de base cc-pVDZ.
Étapes 1 à 4
Ici, nous regroupons toutes les étapes dans un seul workflow à plus grande échelle, qui est ensuite exécuté sur du vrai matériel quantique.
# ------------------------------ Step 1 ------------------------------
# Build N2 molecule
mol = pyscf.gto.Mole()
mol.build(
atom=[["N", (0, 0, 0)], ["N", (1.0, 0, 0)]],
basis="cc-pvdz",
symmetry="Dooh",
)
# Define active space
n_frozen = 2
active_space = range(n_frozen, mol.nao_nr())
# Get molecular integrals
scf = pyscf.scf.RHF(mol).run()
norb = len(active_space)
n_electrons = int(sum(scf.mo_occ[active_space]))
n_alpha = (n_electrons + mol.spin) // 2
n_beta = (n_electrons - mol.spin) // 2
nelec = (n_alpha, n_beta)
cas = pyscf.mcscf.CASCI(scf, norb, nelec)
mo = cas.sort_mo(active_space, base=0)
hcore, nuclear_repulsion_energy = cas.get_h1cas(mo)
eri = pyscf.ao2mo.restore(1, cas.get_h2cas(mo), norb)
# Store reference energy from SCI calculation performed separately
reference_energy = -109.22802921665716
print(f"norb = {norb}")
print(f"nelec = {nelec}")
# Get CCSD t2 amplitudes for initializing the ansatz
ccsd = pyscf.cc.CCSD(
scf, frozen=[i for i in range(mol.nao_nr()) if i not in active_space]
).run()
t1 = ccsd.t1
t2 = ccsd.t2
# Set ansatz properties
n_reps = 1
pairs_aa = [(p, p + 1) for p in range(norb - 1)]
# Let generate_lucj_pass_manager determine the alpha-beta interactions
pairs_ab = None
# Initialize backend
service = QiskitRuntimeService()
backend = service.least_busy(
operational=True, simulator=False, min_num_qubits=133
)
print(f"Using backend {backend.name}")
# Create pass manager
pass_manager, pairs_ab = ffsim.qiskit.generate_lucj_pass_manager(
backend=backend,
norb=norb,
connectivity="heavy-hex",
interaction_pairs=(pairs_aa, pairs_ab),
optimization_level=3,
)
# Create the LUCJ ansatz operator
ucj_op = ffsim.UCJOpSpinBalanced.from_t_amplitudes(
t2=t2,
t1=t1,
n_reps=n_reps,
interaction_pairs=(pairs_aa, pairs_ab),
# Setting optimize=True enables the "compressed" factorization
optimize=True,
# Limit the number of optimization iterations to prevent the code cell
# from running too long. Removing this line may improve results.
options=dict(maxiter=1000),
)
# create an empty quantum circuit
qubits = QuantumRegister(2 * norb, name="q")
circuit = QuantumCircuit(qubits)
# prepare Hartree-Fock state as the reference state and append it
# to the quantum circuit
circuit.append(ffsim.qiskit.PrepareHartreeFockJW(norb, nelec), qubits)
# apply the UCJ operator to the reference state
circuit.append(ffsim.qiskit.UCJOpSpinBalancedJW(ucj_op), qubits)
circuit.measure_all()
# ------------------------------ Step 2 ------------------------------
isa_circuit = pass_manager.run(circuit)
print(f"Gate counts: {isa_circuit.count_ops()}")
# ------------------------------ Step 3 ------------------------------
sampler = Sampler(mode=backend)
sampler.options.environment.job_tags = ["TUT_SQD"]
job = sampler.run([isa_circuit], shots=100_000)
primitive_result = job.result()
pub_result = primitive_result[0]
# ------------------------------ Step 4 ------------------------------
bit_array = pub_result.data.meas
num_valid = sum(
is_valid_bitstring(b, norb, nelec) for b in bit_array.get_bitstrings()
)
valid_fraction = num_valid / bit_array.num_shots
print(f"Fraction of sampled configurations that are valid: {valid_fraction}")
expected_fraction_random = (
math.comb(norb, n_alpha) * math.comb(norb, n_beta) / 2 ** (2 * norb)
)
print(
f"Expected fraction of valid configurations from uniformly random bitstrings: "
f"{expected_fraction_random}"
)
# SQD options
energy_tol = 1e-3
occupancies_tol = 1e-3
max_iterations = 5
# Eigenstate solver options
num_batches = 3
samples_per_batch = 300
symmetrize_spin = True
carryover_threshold = 1e-4
max_cycle = 200
# Use the Hartree-Fock configuration as an initial guess for the
# orbital occupancies
initial_occupancies = (
np.array([1] * n_alpha + [0] * (norb - n_alpha)),
np.array([1] * n_beta + [0] * (norb - n_beta)),
)
# Pass options to the built-in eigensolver. If you just want to use the defaults,
# you can omit this step, in which case you would not specify the
# sci_solver argument in the call to diagonalize_fermionic_hamiltonian below.
sci_solver = partial(solve_sci_batch, spin_sq=0.0, max_cycle=max_cycle)
# List to capture intermediate results
result_history = []
result = diagonalize_fermionic_hamiltonian(
hcore,
eri,
bit_array,
samples_per_batch=samples_per_batch,
norb=norb,
nelec=nelec,
num_batches=num_batches,
energy_tol=energy_tol,
occupancies_tol=occupancies_tol,
max_iterations=max_iterations,
sci_solver=sci_solver,
symmetrize_spin=symmetrize_spin,
initial_occupancies=initial_occupancies,
carryover_threshold=carryover_threshold,
callback=callback,
seed=rng,
)
final_energy = result.energy + nuclear_repulsion_energy
energy_error = final_energy - reference_energy
print(f"Final energy: {final_energy}")
print(f"Final energy error: {energy_error}")
# Data for energies plot
x1 = range(len(result_history))
min_e = [
min(result, key=lambda res: res.energy).energy + nuclear_repulsion_energy
for result in result_history
]
e_diff = [abs(e - reference_energy) for e in min_e]
yt1 = [1.0, 1e-1, 1e-2, 1e-3, 1e-4]
# Chemical accuracy (+/- 1 milli-Hartree)
chem_accuracy = 0.001
# Data for avg spatial orbital occupancy
y2 = np.sum(result.orbital_occupancies, axis=0)
x2 = range(len(y2))
fig, axs = plt.subplots(1, 2, figsize=(12, 6))
# Plot energies
axs[0].plot(x1, e_diff, label="energy error", marker="o")
axs[0].set_xticks(x1)
axs[0].set_xticklabels(x1)
axs[0].set_yticks(yt1)
axs[0].set_yticklabels(yt1)
axs[0].set_yscale("log")
axs[0].set_ylim(1e-4)
axs[0].axhline(
y=chem_accuracy,
color="#BF5700",
linestyle="--",
label="chemical accuracy",
)
axs[0].set_title("Approximated Ground State Energy Error vs SQD Iterations")
axs[0].set_xlabel("Iteration Index", fontdict={"fontsize": 12})
axs[0].set_ylabel("Energy Error (Ha)", fontdict={"fontsize": 12})
axs[0].legend()
# Plot orbital occupancy
axs[1].bar(x2, y2, width=0.8)
axs[1].set_xticks(x2)
axs[1].set_xticklabels(x2)
axs[1].set_title("Avg Occupancy per Spatial Orbital")
axs[1].set_xlabel("Orbital Index", fontdict={"fontsize": 12})
axs[1].set_ylabel("Avg Occupancy", fontdict={"fontsize": 12})
plt.tight_layout()
plt.show()
converged SCF energy = -108.929838385609
norb = 26
nelec = (5, 5)
E(CCSD) = -109.2177884185544 E_corr = -0.2879500329450045
Using backend ibm_boston
Warning: Backend cannot accommodate pairs_ab=[(0, 0), (4, 4), (8, 8), (12, 12), (16, 16), (20, 20), (24, 24)].
Removing interaction (24, 24) from the end.
Warning: Backend cannot accommodate pairs_ab=[(0, 0), (4, 4), (8, 8), (12, 12), (16, 16), (20, 20)].
Removing interaction (20, 20) from the end.
Gate counts: OrderedDict({'sx': 7039, 'rz': 6990, 'cz': 1858, 'x': 61, 'measure': 52, 'barrier': 1})
Fraction of sampled configurations that are valid: 0.02124
Expected fraction of valid configurations from uniformly random bitstrings: 9.607888706852918e-07
Iteration 1
Subsample 0
Energy: -109.13889134249762
Subspace dimension: 120409
Subsample 1
Energy: -109.11785470455858
Subspace dimension: 110889
Subsample 2
Energy: -109.13234360554011
Subspace dimension: 130321
Iteration 2
Subsample 0
Energy: -109.16392179579177
Subspace dimension: 223729
Subsample 1
Energy: -109.16281938332986
Subspace dimension: 223729
Subsample 2
Energy: -109.16955816711932
Subspace dimension: 233289
Iteration 3
Subsample 0
Energy: -109.17905772999075
Subspace dimension: 324900
Subsample 1
Energy: -109.17532445048462
Subspace dimension: 357604
Subsample 2
Energy: -109.1733168689756
Subspace dimension: 348100
Iteration 4
Subsample 0
Energy: -109.18437778820451
Subspace dimension: 474721
Subsample 1
Energy: -109.18450164209159
Subspace dimension: 476100
Subsample 2
Energy: -109.18493571190754
Subspace dimension: 487204
Iteration 5
Subsample 0
Energy: -109.18616522497996
Subspace dimension: 622521
Subsample 1
Energy: -109.18652868888333
Subspace dimension: 644809
Subsample 2
Energy: -109.18753326484406
Subspace dimension: 585225
Final energy: -109.18753326484406
Final energy error: 0.040495951813099396
Prochaines étapes
Si tu as trouvé ce travail intéressant, tu pourrais être intéressé par le matériel suivant :
- Sample-based Krylov quantum diagonalization of a fermionic lattice model - un tutoriel connexe utilisant des circuits d'évolution temporelle au lieu d'un ansatz variationnel
- Scale SQD chemistry workflows with Dice solver - une page montrant comment utiliser le logiciel Dice plus efficace pour la diagonalisation
- Documentation API de l'extension SQD - référence pour la fonction
diagonalize_fermionic_hamiltonian - Chemistry beyond the scale of exact diagonalization on a quantum-centric supercomputer - l'article sur lequel ce tutoriel est basé