Solveur quantique variationnel des valeurs propres (VQE)
Pour ce module, tu dois disposer d'un environnement Python fonctionnel et avoir installé les dernières versions des packages suivants :
qiskitqiskit_ibm_runtimeqiskit-aerqiskit.visualizationnumpypylatexenc
Pour configurer et installer ces packages, consulte le guide Installer Qiskit. Pour exécuter des tâches sur de vrais ordinateurs quantiques, tu devras créer un compte IBM Cloud en suivant les étapes du guide Configurer ton compte IBM Cloud.
Ce module a été testé et a utilisé environ 8 minutes de temps QPU. Il s'agit d'une estimation, et ton utilisation réelle peut varier.
# Added by doQumentation — required packages for this notebook
!pip install -q matplotlib numpy qiskit qiskit-aer qiskit-ibm-runtime scipy
# Uncomment and modify this line as needed to install dependencies
#!pip install 'qiskit>=2.1.0' 'qiskit-ibm-runtime>=0.40.1' 'qiskit-aer>=0.17.0' 'numpy' 'pylatexenc'
Introduction
Depuis le développement du modèle mécanique quantique au début du XXe siècle, les scientifiques ont compris que les électrons ne suivent pas des trajectoires fixes autour du noyau d'un atome, mais existent plutôt dans des régions de probabilité appelées orbitales. Ces orbitales correspondent à des niveaux d'énergie spécifiques et discrets que les électrons peuvent occuper. Les électrons résident naturellement dans les niveaux d'énergie les plus bas disponibles, connus sous le nom d'état fondamental. Cependant, si un électron absorbe suffisamment d'énergie, il peut sauter à un niveau d'énergie supérieur, entrant dans un état excité. Cet état excité est temporaire, et l'électron finira par revenir à un niveau d'énergie inférieur, libérant l'énergie absorbée, souvent sous forme de lumière. Ce processus fondamental d'absorption et d'émission d'énergie est essentiel pour comprendre comment les atomes interagissent et forment des liaisons.
Lorsque des atomes se réunissent pour former des molécules, leurs orbitales atomiques se combinent pour former des orbitales moléculaires. L'arrangement et les niveaux d'énergie des électrons au sein de ces orbitales moléculaires dictent les propriétés de la molécule résultante et la force des liaisons chimiques. Par exemple, lors de la formation d'une molécule d'hydrogène () à partir de deux atomes d'hydrogène individuels, l'électron de chaque atome occupe des orbitales atomiques. À mesure que les atomes se rapprochent, ces orbitales atomiques se chevauchent et se combinent pour former de nouvelles orbitales moléculaires — l'une à énergie plus basse (une orbitale liante) et l'autre à énergie plus haute (une orbitale antiliante). Les deux électrons, un de chaque atome d'hydrogène, occuperont de préférence l'orbitale liante à plus basse énergie, conduisant à la formation d'une liaison covalente stable qui maintient la molécule ensemble. La différence d'énergie entre les atomes séparés et la molécule formée, en particulier l'énergie des électrons dans les orbitales moléculaires, détermine la stabilité et les propriétés de la liaison.
Dans les sections suivantes, nous allons explorer ce processus de formation moléculaire, en nous concentrant sur la molécule . Nous utiliserons un vrai ordinateur quantique, combiné à des techniques d'optimisation classiques, pour trouver l'énergie de ce processus simple mais fondamental. Cette expérience fournira une démonstration pratique de la façon dont le calcul quantique peut être appliqué pour résoudre des problèmes en chimie computationnelle, en apportant des éclairages sur le rôle de l'énergie des électrons.
VQE - Un algorithme quantique variationnel pour les problèmes aux valeurs propres
Techniques d'approximation en chimie - principe variationnel et ensemble de base
Les contributions d'Erwin Schrödinger à la mécanique quantique ne se limitent pas à l'introduction d'un nouveau modèle électronique ; fondamentalement, il a établi la mécanique ondulatoire en développant la célèbre équation de Schrödinger dépendante du temps :
Ici, est l'opérateur hamiltonien, qui représente l'énergie totale du système, et est la fonction d'onde qui contient toutes les informations sur l'état quantique du système. (Note : est la dérivée totale par rapport au temps, et nous n'incluons pas explicitement la valeur propre d'énergie ici.)
Cependant, dans de nombreuses applications pratiques — comme la détermination des niveaux d'énergie autorisés des atomes et des molécules — nous utilisons plutôt l'équation de Schrödinger indépendante du temps (équation aux valeurs propres d'énergie), qui est dérivée de la forme dépendante du temps en supposant un état stationnaire. Un état stationnaire est un état quantique dans lequel la densité de probabilité de trouver une particule en un point donné de l'espace ne change pas au fil du temps.
Dans cette forme, représente la valeur propre d'énergie correspondant à l'état quantique . L'hamiltonien inclut diverses contributions énergétiques, comme l'énergie cinétique des électrons et des noyaux, les forces attractives entre électrons et noyaux, et les forces répulsives entre électrons.
Résoudre l'équation aux valeurs propres d'énergie nous permet de calculer les niveaux d'énergie quantifiés des systèmes atomiques et moléculaires. Cependant, pour les molécules, la résoudre exactement est difficile car la fonction d'onde , qui décrit la distribution spatiale des électrons, est complexe et de haute dimension.
En conséquence, les scientifiques utilisent des techniques d'approximation pour obtenir des solutions pratiques et précises. Dans ce travail, nous nous concentrerons sur deux méthodes clés :
-
Principe variationnel
Cette méthode approxime la fonction d'onde et l'ajuste pour se rapprocher le plus possible de l'énergie cible, généralement l'énergie de l'état fondamental du système. L'idée centrale du principe variationnel est simple :
- Si nous devinons une fonction d'onde (une « fonction d'essai »), l'énergie calculée à partir de celle-ci sera toujours égale ou supérieure à l'énergie de l'état fondamental () du système.
- En ajustant les paramètres dans la fonction d'essai, , on peut obtenir une approximation de plus en plus précise de l'énergie de l'état fondamental.
- Sa précision dépend fortement du choix de la fonction d'onde d'essai . Une fonction d'essai mal choisie peut conduire à une estimation d'énergie éloignée de la réalité.
-
Approximation par ensemble de base
La deuxième méthode d'approximation intervient lors de la construction de la fonction d'onde — l'approche par ensemble de base. En chimie quantique, résoudre exactement l'équation de Schrödinger pour les molécules est pratiquement impossible. À la place, nous approximons la fonction d'onde complexe à multi-électrons en la construisant à partir de fonctions mathématiques plus simples et prédéfinies. Un ensemble de base est essentiellement une collection de ces fonctions mathématiques connues, typiquement centrées sur les atomes de la molécule, utilisées comme briques de construction pour représenter la forme et le comportement des électrons dans le système. Imagine que tu essaies de recréer une sculpture détaillée en utilisant uniquement une collection de briques LEGO standard — plus tu as de types et de tailles de briques (plus l'ensemble de base est grand), plus tu peux approximer précisément la forme originale.
Ces fonctions de base sont souvent inspirées des solutions analytiques pour des systèmes simples comme l'atome d'hydrogène, prenant des formes comme les fonctions gaussiennes ou de type Slater, bien qu'elles restent des approximations. Au lieu de travailler avec les orbitales moléculaires complètes théoriquement « exactes » mais inextricables, nous les exprimons comme une combinaison linéaire (une somme avec des coefficients) de ces fonctions de base. Cette méthode est connue sous le nom d'approche LCAO (Combinaison Linéaire d'Orbitales Atomiques) lorsque les fonctions de base ressemblent à des orbitales atomiques. En optimisant les coefficients dans cette combinaison linéaire, nous pouvons trouver la meilleure fonction d'onde et énergie approximatives possibles dans les limites de l'ensemble de base choisi.
- Plus il y a de fonctions incluses dans l'ensemble de base, meilleure est l'approximation, mais cela se fait au prix d'un effort de calcul plus élevé.
- Un petit ensemble de base fournit une estimation grossière, tandis qu'un grand ensemble de base donne des résultats plus précis au prix de ressources de calcul plus importantes.
Pour résumer, pour rendre les calculs réalisables et réduire les coûts de calcul, nous utilisons le principe variationnel en approximant la fonction d'onde, ce qui réduit la complexité calculatoire et permet une optimisation itérative pour minimiser l'énergie. Par ailleurs, l'approche par ensemble de base simplifie les calculs en représentant les orbitales atomiques comme une combinaison de fonctions prédéfinies, plutôt que de résoudre directement une fonction d'onde continue.
Teste ta compréhension
Considère la fonction d'onde d'essai où est une constante de normalisation et est un paramètre ajustable.
(a) Normalise la fonction d'onde d'essai en déterminant tel que .
Answer
Pour normaliser la fonction d'onde d'essai donnée :
On utilise l'intégrale gaussienne :
On pose et on obtient :
(b) Calcule la valeur d'espérance de l'hamiltonien donné par où , ce qui correspond à un potentiel d'oscillateur harmonique simple.
Answer
L'hamiltonien pour un oscillateur harmonique est :
Valeur d'espérance de l'énergie cinétique
En calculant la dérivée seconde :
Ainsi :
En utilisant les résultats standards des intégrales gaussiennes :
Valeur d'espérance de l'énergie potentielle
En utilisant :
on obtient :
Valeur d'espérance de l'énergie totale
(c) Utilise le principe variationnel pour trouver l' optimal en minimisant .
Answer
Optimisation de pour l'énergie minimale
On dérive
En résolvant :
En substituant dans :
ce qui correspond exactement à l'énergie de l'état fondamental de l'oscillateur harmonique quantique.
VQE (Solveur quantique variationnel des valeurs propres)
Le solveur quantique variationnel des valeurs propres (VQE) est la méthode principale que nous utiliserons pour explorer le processus , et ici nous allons examiner ce qu'est le VQE et comment il fonctionne. Mais faisons d'abord une pause et regardons une chose très importante à travers la question de vérification.
Teste ta compréhension
Si nous disposons déjà d'autant de stratégies pour les problèmes de chimie, pourquoi avons-nous besoin d'un ordinateur quantique ? Et quel est l'intérêt d'utiliser à la fois des ordinateurs quantiques et classiques ensemble ?
Answer
L'informatique quantique a la chance de révolutionner la chimie en s'attaquant à des problèmes que les ordinateurs classiques peinent à résoudre en raison de la mise à l'échelle exponentielle des états quantiques. Richard Feynman a célèbrement noté que pour simuler la nature, les calculs doivent également être quantiques [réf. 1].
Par exemple, simuler la caféine avec l'ensemble de base le plus simple (STO-3G) nécessiterait bits, bien plus grand que le nombre total d'étoiles dans l'univers observable () [réf. 2]. Un ordinateur quantique peut décrire les orbitales électroniques de la caféine avec 160 qubits.
Les ordinateurs quantiques traitent naturellement les interactions quantiques en utilisant la superposition et l'intrication, ce qui offre une voie prometteuse pour permettre des simulations moléculaires précises. De plus, nous pouvons combiner les avantages des ordinateurs quantiques (simulation électronique) et classiques (pré/post-traitement des données, gestion des processus algorithmiques, optimisation, etc.). Ces approches devraient améliorer la découverte de matériaux, la conception de médicaments et les prédictions de réactions, réduisant ainsi les coûts des expériences par essais et erreurs. [réf. 3][réf. 4]
Si tu veux savoir pourquoi les ordinateurs quantiques sont nécessaires pour les problèmes de chimie et pourquoi utiliser à la fois des ressources de calcul quantiques et classiques, consulte les articles suivants :
Revenons maintenant au VQE.
Le VQE combine la puissance des ordinateurs quantiques et des ordinateurs classiques, en utilisant fondamentalement des principes variationnels pour obtenir l'énergie de l'état fondamental du système. Pour comprendre le VQE, décompose-le en trois parties :
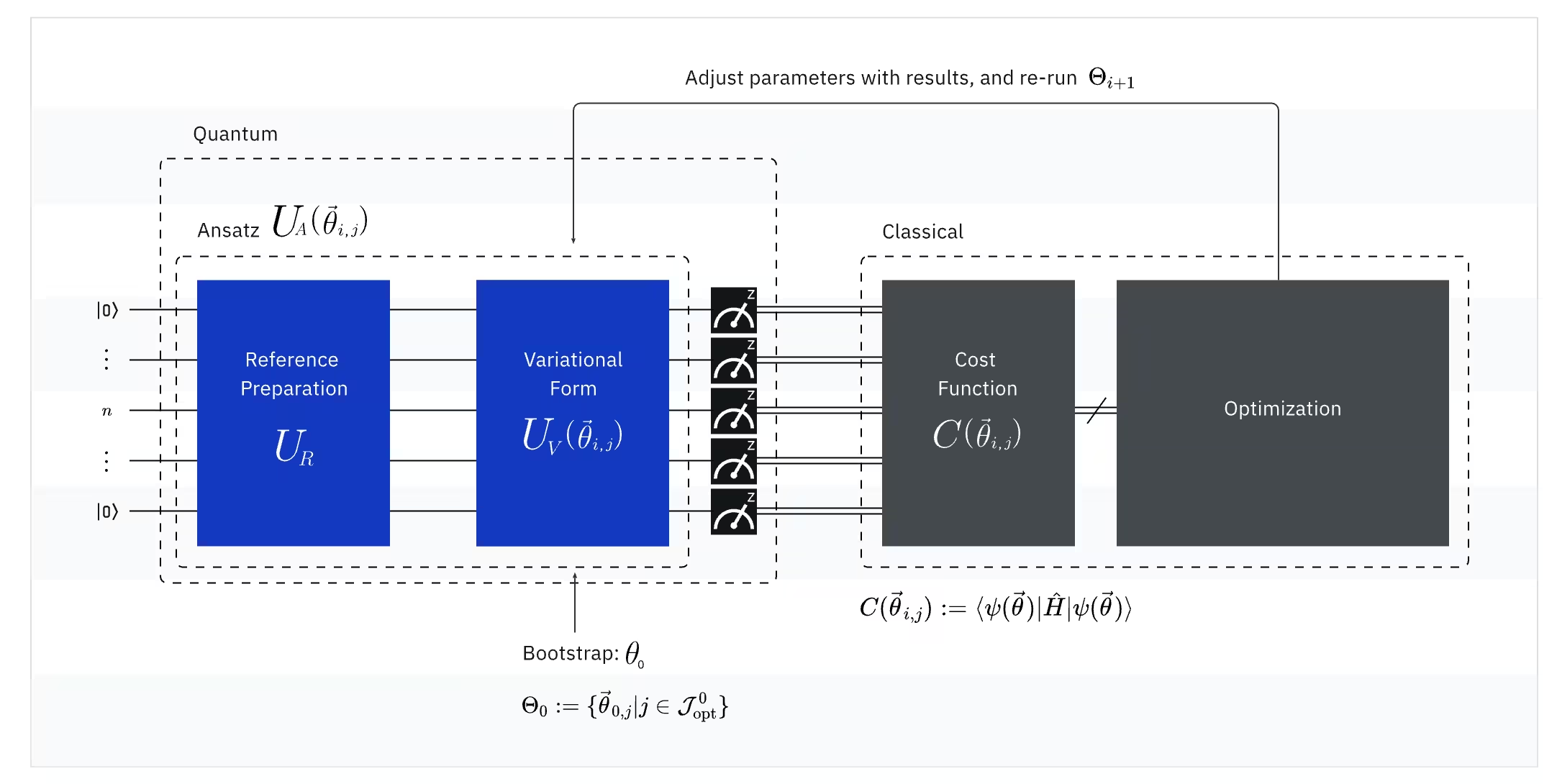
(Quantique) Observable : l'hamiltonien moléculaire (énergie d'une molécule)
Dans le VQE, l'hamiltonien moléculaire/atomique est une observable, ce qui signifie que nous pouvons mesurer sa valeur à travers une expérience. Notre objectif est de trouver l'énergie la plus basse possible (l'énergie de l'état fondamental) de la molécule. Pour ce faire, nous utilisons un état quantique d'essai, généré par un circuit quantique paramétré (ansatz). Nous mesurons l'observable et optimisons l'état quantique jusqu'à atteindre l'énergie la plus basse possible.
L'ensemble de base utilisé pour l'hamiltonien moléculaire détermine le nombre de qubits requis et affecte directement la précision du VQE. Choisir le bon ensemble de base est essentiel pour équilibrer efficacité et précision. Pour simplifier les calculs sans changer l'ensemble de base, nous pouvons utiliser des stratégies comme l'imposition de symétrie et la réduction de l'espace actif. De nombreuses molécules ont des formes symétriques (comme un papillon ou un flocon de neige), ce qui signifie que certaines parties se comportent de la même façon. Au lieu de tout calculer séparément, nous pouvons nous concentrer uniquement sur les parties uniques, économisant des ressources quantiques et exploitant ainsi la symétrie. Dans la réduction de l'espace actif, nous ne considérons que les orbitales importantes, car tous les électrons n'ont pas d'impact significatif sur l'énergie moléculaire. Les électrons proches du noyau restent pour la plupart inchangés, tandis que d'autres influencent les liaisons. En appliquant ces méthodes, nous pouvons rendre le VQE plus efficace tout en maintenant la précision.
Une fois que nous obtenons un hamiltonien moléculaire en utilisant l'ensemble de base approprié et les stratégies ci-dessus, nous devons transformer cet hamiltonien en un hamiltonien adapté aux ordinateurs quantiques. La mise en correspondance des problèmes avec les opérateurs de Pauli peut être assez compliquée. C'est particulièrement vrai en chimie quantique, qui traite des particules indiscernables (électrons), alors que les qubits sont discernables. Nous n'entrerons pas dans les détails des correspondances ici, mais nous te renvoyons aux ressources suivantes. Une discussion générale sur la mise en correspondance d'un problème avec des opérateurs quantiques se trouve dans Quantum computing in practice. Une discussion plus détaillée sur la mise en correspondance des problèmes de chimie en opérateurs quantiques se trouve dans Quantum chemistry with VQE.
Pour ce module, nous te fournirons les hamiltoniens (à un qubit) appropriés pour et afin que nous puissions nous concentrer sur l'utilisation de l'ordinateur quantique. Ces hamiltoniens à un qubit sont préparés en utilisant l'ensemble de base STO-6G et la correspondance de Jordan-Wigner, qui est la correspondance la plus directe avec l'interprétation physique la plus simple, car elle met en correspondance l'occupation d'une spin-orbitale avec l'occupation d'un qubit. Nous avons également utilisé une technique de réduction de qubits en exploitant une symétrie de l'hamiltonien, qui utilise les motifs dans le comportement des occupations de spin pour réduire le nombre de qubits. Pour la molécule , nous supposons que la distance entre les deux atomes d'hydrogène est 0.735 .
(Quantique) Ansatz : la fonction d'onde d'essai (comment construire un état quantique d'essai avec un circuit quantique)
Pour le VQE, l'ansatz (pluriel : ansätze) se compose de deux composantes clés. La première est la préparation de l'état initial, qui configure l'état du qubit en appliquant des portes quantiques sans paramètre variationnel. La deuxième composante est le circuit quantique paramétré, un circuit quantique spécial avec des paramètres ajustables, semblable aux boutons d'une radio. Ces paramètres seront utilisés par la dernière partie — l'optimiseur classique — pour nous aider à atteindre le meilleur état fondamental possible.
Dans la section sur le principe variationnel, nous avons appris que la qualité de l'état d'essai affecte la qualité des résultats de l'algorithme variationnel. Cela signifie que choisir un bon ansatz est important dans le VQE. Encore une fois, il s'agit d'un sujet riche et complexe. Nous ne couvrirons pas ici les différents types d'ansatz ni leurs origines. Si tu souhaites en savoir plus sur les circuits quantiques paramétrés et les ansatz, tu peux explorer la leçon Ansatz and variational form du cours sur la conception d'algorithmes variationnels, qui fournit des explications et des exemples détaillés d'ansätze.
Puisque nous allons utiliser un hamiltonien à un qubit dans ce module, nous avons besoin d'un circuit quantique paramétré à un qubit comme ansatz. Nous verrons trois types d'ansätze à un qubit dans la section suivante. Nous les comparerons et discuterons des considérations clés dans le choix d'un ansatz.
(Classique) Optimiseur : ajustement fin du circuit quantique
Une fois que l'ordinateur quantique mesure l'énergie de l'observable à partir de l'ansatz, les paramètres de l'ansatz et la valeur d'énergie sont envoyés à l'optimiseur classique pour ajustement. Ce processus d'optimisation est effectué sur un ordinateur classique, généralement à l'aide de packages scientifiques polyvalents comme SciPy.
L'optimiseur classique traite l'énergie mesurée comme une fonction de coût. Dans les problèmes d'optimisation, une fonction de coût (aussi parfois appelée fonction objectif) est une fonction mathématique qui mesure à quel point une solution particulière est « bonne ». L'objectif de l'optimiseur est de trouver l'ensemble de paramètres qui minimise cette fonction de coût. Dans le contexte de la recherche de l'énergie de l'état fondamental d'une molécule, l'énergie elle-même sert de fonction de coût — nous voulons trouver les paramètres de notre circuit quantique (notre « solution ») qui donnent l'énergie la plus basse possible. L'optimiseur classique utilise cette valeur d'énergie mesurée (le coût) et détermine le prochain ensemble de paramètres optimisés pour l'ansatz quantique. Ces paramètres mis à jour sont ensuite renvoyés au circuit quantique, et le processus se répète. À chaque itération, l'optimiseur classique ajuste les paramètres pour tenter de réduire l'énergie (minimiser la fonction de coût) jusqu'à ce qu'un critère de convergence prédéfini soit atteint, garantissant idéalement que l'énergie la plus basse possible (correspondant à l'état fondamental de la molécule pour cette distance de liaison et cet ensemble de base) est trouvée.
Il existe de nombreuses stratégies d'optimisation fournies par des packages scientifiques comme SciPy. Tu peux en trouver davantage dans la leçon Optimization loops du cours Variational algorithm design. Ici, nous utiliserons COBYLA (Constrained Optimization BY Linear Approximations), un algorithme d'optimisation adapté aux paysages énergétiques complexes. En particulier, COBYLA ne tente pas de calculer un gradient de la fonction étudiée ; on appelle cela un optimiseur sans gradient. Imagine que tu essaies de trouver le pic le plus haut dans une chaîne de montagnes les yeux fermés. Comme tu ne peux pas voir tout le paysage, tu fais de petits pas dans différentes directions, en vérifiant si tu montes ou descends. COBYLA fonctionne de manière similaire — il se déplace dans l'espace des paramètres, testant différentes valeurs, améliorant progressivement le résultat jusqu'à trouver le meilleur.
Tu es maintenant prêt·e à effectuer un calcul VQE. Pour ce faire, essaie la question de vérification ci-dessous, qui récapitule le processus global.
Teste ta compréhension
Remplis les blancs avec les termes corrects pour compléter le résumé du processus VQE, puis clique pour vérifier tes réponses.
Le VQE est un algorithme quantique variationnel, qui combine la puissance de (1) ________ et de l'informatique classique, utilisé pour trouver (2) __________ d'une molécule. Le processus commence par la définition de (3) __________, qui représente l'énergie totale du système et agit comme l'observable dans les mesures quantiques. Ensuite, nous préparons un (4) __________, un circuit quantique avec des paramètres ajustables qui représente la fonction d'onde d'essai de la molécule. Ces paramètres sont optimisés à l'aide d'un (5) __________, un algorithme classique qui ajuste les paramètres de manière itérative pour minimiser l'énergie mesurée. Dans la discussion ci-dessus, nous avons utilisé l'optimiseur (6) __________, qui affine les paramètres de l'ansatz sans avoir besoin de calculs de dérivées. Le processus continue jusqu'à ce que nous atteignions (7) __________, ce qui signifie que nous avons trouvé l'énergie la plus basse possible de la molécule.
Banque de mots :
- optimiseur classique
- énergie de l'état fondamental
- efficace sur le matériel
- ansatz
- hamiltonien moléculaire
- COBYLA
- informatique quantique
- convergence
Answer
1 → informatique quantique
2 → énergie de l'état fondamental
3 → hamiltonien moléculaire
4 → ansatz
5 → optimiseur classique
6 → COBYLA
7 → convergence
Calculer l'énergie de l'état fondamental d'un atome d'hydrogène avec VQE
Maintenant, utilisons ce que nous avons appris pour calculer l'énergie de l'état fondamental d'un atome d'hydrogène. Tout au long du module, nous utiliserons un cadre de travail pour le calcul quantique appelé « Qiskit patterns » (motifs Qiskit), qui décompose les flux de travail en étapes suivantes :
- Étape 1 : Transformer les entrées classiques en un problème quantique
- Étape 2 : Optimiser le problème pour l'exécution quantique
- Étape 3 : Exécuter avec les primitives Qiskit Runtime
- Étape 4 : Post-traitement et analyse classique

Nous suivrons ces étapes dans l'ensemble.
Commençons par charger les paquets nécessaires, y compris les primitives Qiskit Runtime. Nous sélectionnerons également l'ordinateur quantique le moins occupé disponible.
Le code ci-dessous permet de sauvegarder tes identifiants lors de la première utilisation. Pense bien à supprimer ces informations du notebook après les avoir enregistrées dans ton environnement, afin que tes identifiants ne soient pas partagés accidentellement lorsque tu partages le notebook. Consulte Configurer ton compte IBM Cloud et Initialiser le service dans un environnement non fiable pour plus d'informations.
# Load the Qiskit Runtime service
from qiskit_ibm_runtime import QiskitRuntimeService
# Load the Runtime primitive and session
from qiskit_ibm_runtime import EstimatorV2 as Estimator
# Syntax for first saving your token. Delete these lines after saving your credentials.
# QiskitRuntimeService.save_account(channel='ibm_quantum_platform',
# instance = '<YOUR_IBM_INSTANCE_CRN>', token='<YOUR-API_KEY>', overwrite=True, set_as_default=True)
# service = QiskitRuntimeService(channel='ibm_quantum_platform')
# Load saved credentials
service = QiskitRuntimeService()
# Use the least busy backend, or uncomment the loading of a specific backend like "ibm_brisbane".
backend = service.least_busy(operational=True, simulator=False, min_num_qubits=127)
# backend = service.backend("ibm_brisbane")
print(backend.name)
ibm_brisbane
La cellule ci-dessous te permet de basculer entre le simulateur et le vrai matériel tout au long du notebook. Nous te recommandons de l'exécuter maintenant :
# Load the Aer simulator and generate a noise model based on the currently-selected backend.
from qiskit_aer import AerSimulator
from qiskit_aer.noise import NoiseModel
# Alternatively, load a fake backend with generic properties and define a simulator.
noise_model = NoiseModel.from_backend(backend)
# Define a simulator using Aer, and use it in Sampler.
backend_sim = AerSimulator(noise_model=noise_model)
Étape 1 : Transformer le problème en circuits quantiques et en opérateurs
Nous commençons notre calcul VQE en définissant l'Hamiltonien de la molécule d'hydrogène () à une distance de liaison spécifique. Cet Hamiltonien représente l'énergie totale du système en termes d'opérateurs de qubits, ayant été produit et mappé à partir du système moléculaire à l'aide d'une procédure standard : 1) utilisation de l'ensemble de base STO-6G (un ensemble spécifique de fonctions mathématiques servant à approximer les orbitales électroniques), 2) application du mapping Jordan-Wigner (une technique pour traduire les opérateurs fermioniques décrivant les électrons en opérateurs de qubits), et 3) réduction du nombre de qubits à l'aide de la parité de l'Hamiltonien pour simplifier le problème.
Comme nous l'avons expliqué précédemment, les énergies de l'état fondamental calculées dépendent fortement du choix de l'ensemble de base et de la géométrie moléculaire (comme la distance de liaison). Pour cette configuration spécifique et après ces transformations, l'Hamiltonien de qubits résultant est simple :
Ici, représente l'opérateur identité et représente l'opérateur de Pauli-Z, agissant sur un seul qubit. Les coefficients sont dérivés des intégrales calculées avec l'ensemble de base STO-6G à cette distance de liaison particulière, avec la transformation appropriée.
Avec cet Hamiltonien défini, nous pouvons maintenant utiliser VQE pour calculer son énergie de l'état fondamental. Il est utile de comparer notre énergie de l'état fondamental calculée à des valeurs de référence. Pour un atome d'hydrogène (H) isolé, l'énergie de l'état fondamental est exactement -0,5 Hartree (en l'absence d'effets relativistes). Calculons l'énergie exacte de l'état fondamental de notre Hamiltonien de qubits spécifique tel que défini ci-dessus, et comparons-la aux valeurs connues pertinentes.
from qiskit.quantum_info import SparsePauliOp
import numpy as np
# Qubit Hamiltonian of the hydrogen atom generated by using STO-3G basis set and parity mapping
Hamiltonian = SparsePauliOp.from_list([("I", -0.2355), ("Z", 0.2355)])
# exact ground state energy of Hamiltonian
A = np.array(Hamiltonian)
eigenvalues, eigenvectors = np.linalg.eig(A)
print(
"The exact ground state energy of the Hamiltonian is ",
min(eigenvalues).real,
"hartree",
)
h = min(eigenvalues.real)
The exact ground state energy of the Hamiltonian is -0.471 hartree
Ensuite, nous avons besoin d'un circuit quantique paramétré, un ansatz, pour préparer une fonction d'onde d'essai pour l'état fondamental. L'objectif est de trouver les paramètres qui minimisent la valeur d'espérance de l'énergie . Le choix de l'ansatz est crucial car il détermine l'ensemble des états quantiques possibles que notre circuit peut préparer. Un « bon » ansatz est suffisamment flexible pour représenter un état très proche du vrai état fondamental de l'Hamiltonien étudié, sans pour autant être si complexe qu'il nécessite trop de paramètres ou un circuit trop profond pour les ordinateurs quantiques actuels.
Ici, nous allons essayer trois ansätze à un qubit différents pour voir lequel offre une meilleure « couverture » des états quantiques possibles d'un qubit. La « couverture » désigne la plage d'états quantiques que le circuit ansatz peut produire en faisant varier ses paramètres.
Nous utiliserons trois ansätze basés sur différentes combinaisons de portes de rotation à un qubit :
- Un ansatz à une porte de rotation sur un axe : Cet ansatz utilise des rotations autour d'un seul axe (). Sur la sphère de Bloch, cela correspond à se déplacer uniquement le long d'un cercle spécifique. C'est le moins flexible et il couvre un ensemble limité d'états.
- Deux ansätze à portes de rotation sur deux axes : Ces ansätze combinent des rotations autour de deux axes différents ( et ). Cela permet d'atteindre une plus grande portion de la sphère de Bloch, comparé à une rotation sur un seul axe.
En comparant les résultats VQE obtenus avec ces trois ansätze, nous pouvons voir comment la flexibilité et la couverture de l'espace des états de l'ansatz influencent notre capacité à trouver la vraie énergie de l'état fondamental de notre Hamiltonien simplifié. Un ansatz plus flexible a le potentiel de trouver une meilleure approximation, mais peut aussi s'avérer plus difficile à optimiser pour l'optimiseur classique.
from qiskit import QuantumCircuit
from qiskit.circuit import Parameter
from qiskit.quantum_info import Statevector, DensityMatrix, Pauli
theta = Parameter("θ")
phi = Parameter("φ")
lam = Parameter("λ")
ansatz1 = QuantumCircuit(1)
ansatz1.rx(theta, 0)
ansatz2 = QuantumCircuit(1)
ansatz2.rx(theta, 0)
ansatz2.rz(phi, 0)
ansatz3 = QuantumCircuit(1)
ansatz3.rx(theta, 0)
ansatz3.rz(phi, 0)
ansatz3.rx(lam, 0)
<qiskit.circuit.instructionset.InstructionSet at 0x1059def80>
Maintenant, générons 5000 nombres aléatoires pour chaque paramètre et traçons la distribution des états quantiques aléatoires produits par les trois ansätze avec ces paramètres aléatoires. Tu peux considérer ces paramètres comme des rotations autour de différents axes sur une surface sphérique. Pour visualiser la distribution des états quantiques, nous utiliserons la sphère de Bloch, une sphère tridimensionnelle qui représente l'état d'un qubit. Chaque point sur la sphère représente un état possible du qubit, où les pôles nord et sud correspondent aux valeurs classiques « 0 » et « 1 », mais le qubit peut aussi se trouver n'importe où entre les deux, exhibant des propriétés quantiques particulières comme la superposition. Commence par préparer les fonctions nécessaires pour tracer la sphère de Bloch 3D et générer 5000 paramètres aléatoires.
import matplotlib.pyplot as plt
def plot_bloch(bloch_vectors):
# Extract X, Y, Z coordinates for 3D projection
X_coords = bloch_vectors[:, 0]
Z_coords = bloch_vectors[:, 2]
# Compute Y coordinates from X and Z to approximate the full Bloch sphere projection
Y_coords = bloch_vectors[:, 1]
# Create 3D plot
fig = plt.figure(figsize=(8, 8))
ax = fig.add_subplot(111, projection="3d")
ax.scatter(X_coords, Y_coords, Z_coords, color="blue", alpha=0.6)
# Labels and title
ax.set_xlabel("X")
ax.set_ylabel("Y")
ax.set_zlabel("Z")
ax.set_title("Parameterized 1-Qubit Circuit on 3D Bloch Sphere")
# Set axis limits and make them equal
ax.set_xlim([-1, 1])
ax.set_ylim([-1, 1])
ax.set_zlim([-1, 1])
# Ensure equal aspect ratio for all axes
ax.set_box_aspect([1, 1, 1]) # Equal scaling for x, y, z axes
# Show grid
ax.grid(True)
plt.show()
num_samples = 5000 # Number of random states
theta_vals = np.random.uniform(0, 2 * np.pi, num_samples)
phi_vals = np.random.uniform(0, 2 * np.pi, num_samples)
lam_vals = np.random.uniform(0, 2 * np.pi, num_samples)
Voyons comment notre premier ansatz fonctionne.
# List to store Bloch Sphere XZ coordinates
bloch_vectors = []
# Generate quantum states and extract Bloch vectors
for i in range(num_samples):
# Create a circuit and bind parameters
qc = ansatz1
bound_qc = qc.assign_parameters({theta: theta_vals[i]}) # , lam: lam_vals[i]})
state = Statevector.from_instruction(bound_qc)
rho = DensityMatrix(state)
X = rho.expectation_value(Pauli("X")).real
Y = rho.expectation_value(Pauli("Y")).real
Z = rho.expectation_value(Pauli("Z")).real
bloch_vectors.append([X, Y, Z]) # Store X, Z components
# Convert to a numpy array for plotting
bloch_vectors = np.array(bloch_vectors)
plot_bloch(bloch_vectors)
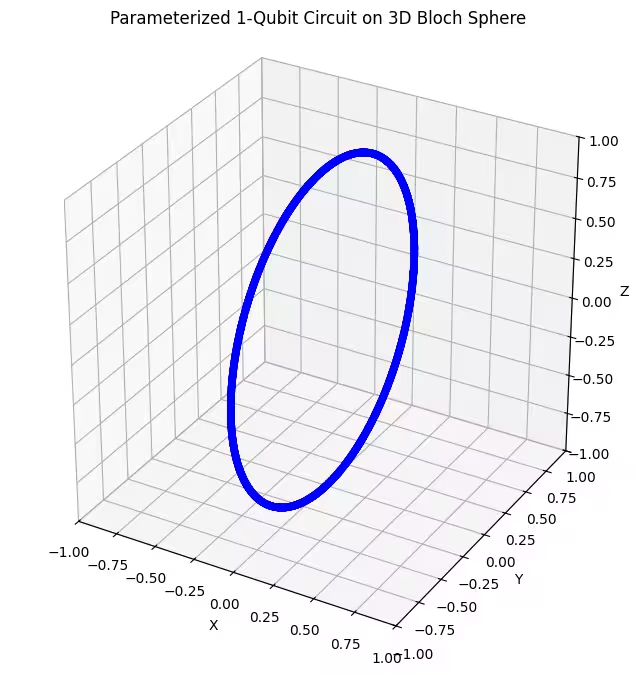
On constate que notre premier ansatz produit des états quantiques disposés en anneau sur la sphère de Bloch. C'est logique, car nous n'avons fourni à l'ansatz qu'un seul paramètre de rotation. Il ne peut donc produire que des états tournés autour d'un seul axe. En partant du point et en tournant autour d'un axe, on obtient toujours un anneau. Vérifions maintenant notre deuxième ansatz, qui comporte deux portes de rotation orthogonales — Rx et Rz.
bloch_vectors = []
# Generate quantum states and extract Bloch vectors
for i in range(num_samples):
# Create circuit and bind parameters
qc = ansatz2
bound_qc = qc.assign_parameters(
{theta: theta_vals[i], phi: phi_vals[i]}
) # , lam: lam_vals[i]})
state = Statevector.from_instruction(bound_qc)
rho = DensityMatrix(state)
X = rho.expectation_value(Pauli("X")).real
Y = rho.expectation_value(Pauli("Y")).real
Z = rho.expectation_value(Pauli("Z")).real
bloch_vectors.append([X, Y, Z]) # Store X, Z components
# Convert to numpy array for plotting
bloch_vectors = np.array(bloch_vectors)
plot_bloch(bloch_vectors)
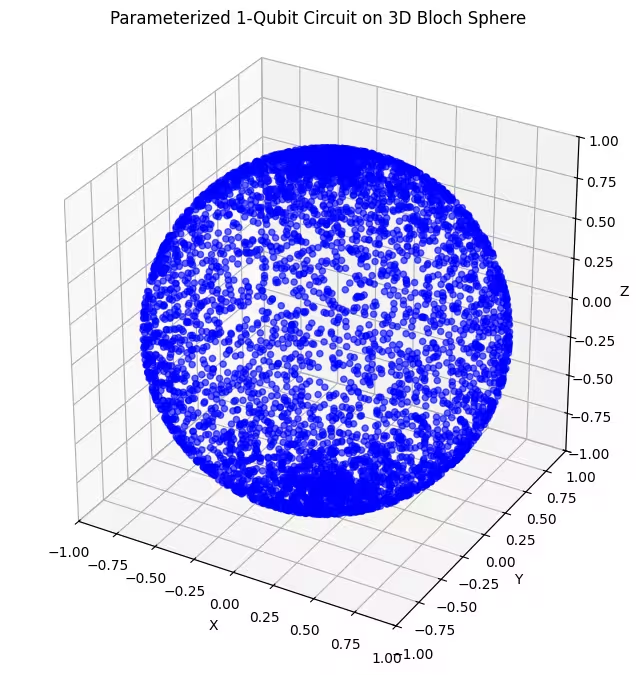
Ici, on voit que notre deuxième ansatz couvre une plus grande portion de la sphère de Bloch — mais note que les points sont plus concentrés autour des pôles et plus dispersés autour de l'équateur. Il est maintenant temps de vérifier notre dernier ansatz.
bloch_vectors = []
# Generate quantum states and extract Bloch vectors
for i in range(num_samples):
# Create circuit and bind parameters
qc = ansatz3
bound_qc = qc.assign_parameters(
{theta: theta_vals[i], phi: phi_vals[i], lam: lam_vals[i]}
)
state = Statevector.from_instruction(bound_qc)
rho = DensityMatrix(state)
X = rho.expectation_value(Pauli("X")).real
Y = rho.expectation_value(Pauli("Y")).real
Z = rho.expectation_value(Pauli("Z")).real
bloch_vectors.append([X, Y, Z]) # Store X, Z components
# Convert to numpy array for plotting
bloch_vectors = np.array(bloch_vectors)
plot_bloch(bloch_vectors)
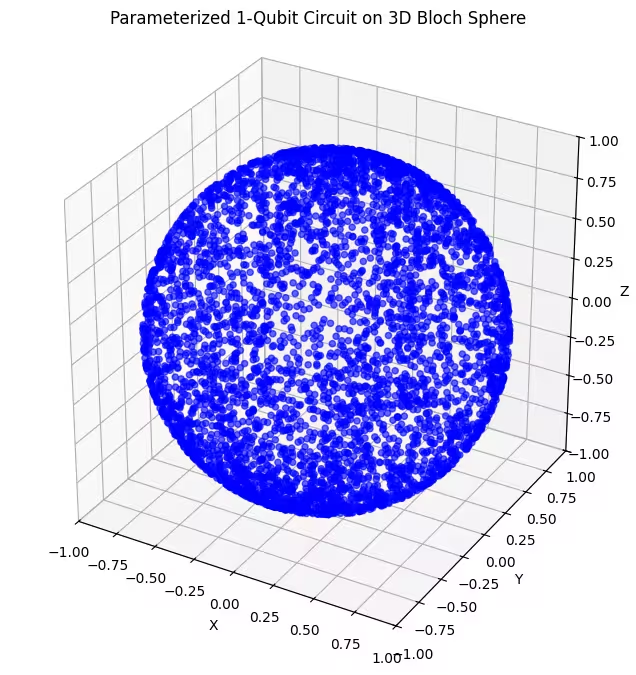
On peut voir ici des états quantiques répartis plus uniformément, générés par notre dernier ansatz.
Comme mentionné, la meilleure approche consiste à s'informer sur l'état fondamental recherché et à utiliser un ansatz bien adapté pour sonder les états proches de cet état fondamental. Par exemple, si nous savions que notre état fondamental se trouvait près d'un pôle, nous pourrions choisir l'ansatz 2. Par souci de simplicité, nous conserverons l'ansatz 3, qui sonde uniformément l'ensemble de la sphère de Bloch.
Maintenant que nous avons sélectionné notre ansatz, traçons le circuit.
# Pre-defined ansatz circuit and operator class for Hamiltonian
ansatz = ansatz3
num_params = ansatz.num_parameters
print("This circuit has ", num_params, "parameters")
ansatz.draw("mpl", style="iqp")
This circuit has 3 parameters

Étape 2 : Optimiser pour le matériel cible
Lorsqu'on exécute un calcul sur un vrai ordinateur quantique, la logique du circuit quantique n'est pas la seule chose qui compte. Il faut aussi tenir compte des opérations que cet ordinateur quantique particulier peut effectuer, ainsi que de l'emplacement des qubits utilisés sur la machine. Sont-ils adjacents ? Sont-ils éloignés les uns des autres ? L'étape suivante consiste donc à réécrire notre circuit en utilisant des portes naturelles pour l'ordinateur quantique choisi, en tenant compte de la disposition des qubits. Cela se fait par transpilation — après ce processus, tu peux voir que notre simple ansatz est converti en un ensemble différent de portes, et que nos qubits abstraits sont mappés vers des qubits physiques sur un vrai ordinateur quantique.
from qiskit.transpiler.preset_passmanagers import generate_preset_pass_manager
config = backend.configuration()
print("Backend: {config.backend_name}")
print("Native gates: ", config.supported_instructions, ",")
target = backend.target
pm = generate_preset_pass_manager(target=target, optimization_level=3)
ansatz_isa = pm.run(ansatz)
ansatz_isa.draw(output="mpl", idle_wires=False, style="iqp")
Backend: {config.backend_name}
Native gates: ['ecr', 'id', 'delay', 'measure', 'reset', 'rz', 'sx', 'x'] ,

On peut voir que les portes rx, rz de notre ansatz ont été converties en une série de portes rz, sx, qui sont les portes natives de notre backend. De plus, on voit que notre q0 est maintenant mappé sur le cinquième qubit physique. Nous devons également mapper notre Hamiltonien en fonction de ces changements, comme dans le code suivant :
Hamiltonian_isa = Hamiltonian.apply_layout(layout=ansatz_isa.layout)
Étape 3 : Exécuter sur le matériel cible
Il est maintenant temps d'exécuter notre VQE sur un vrai QPU. Pour cela, nous avons d'abord besoin d'une fonction de coût pour le processus d'optimisation, qui évalue la valeur d'espérance de l'Hamiltonien avec un état quantique généré par l'ansatz. Ne t'inquiète pas ! Tu n'as pas besoin de tout coder toi-même. Nous avons préparé une fonction pour ça, et il te suffit d'exécuter la cellule ci-dessous.
def cost_func(params, ansatz, hamiltonian, estimator):
"""Return estimate of energy from estimator
Parameters:
params (ndarray): Array of ansatz parameters
ansatz (QuantumCircuit): Parameterized ansatz circuit
hamiltonian (SparsePauliOp): Operator representation of Hamiltonian
estimator (EstimatorV2): Estimator primitive instance
cost_history_dict: Dictionary for storing intermediate results
Returns:
float: Energy estimate
"""
pub = (ansatz, [hamiltonian], [params])
result = estimator.run(pubs=[pub]).result()
energy = result[0].data.evs[0]
cost_history_dict["iters"] += 1
cost_history_dict["prev_vector"] = params
cost_history_dict["cost_history"].append(energy)
print(f"Iters. done: {cost_history_dict['iters']} [Current cost: {energy}]")
return energy
Enfin, préparons les paramètres initiaux pour notre ansatz et son processus d'optimisation. Tu peux simplement utiliser des zéros ou des valeurs aléatoires. Nous avons sélectionné des paramètres initiaux ci-dessous, mais n'hésite pas à commenter ou décommenter les lignes dans la cellule pour échantillonner les paramètres aléatoirement, uniformément entre 0 et .
# x0 = np.random.uniform(0, 2*pi, 3)
x0 = [1, 1, 0]
# QPU Est. 2min for ibm_brisbane
from scipy.optimize import minimize
from qiskit_ibm_runtime import Batch
batch = Batch(backend=backend)
cost_history_dict = {
"prev_vector": None,
"iters": 0,
"cost_history": [],
}
estimator = Estimator(mode=batch)
estimator.options.default_shots = 10000
res = minimize(
cost_func,
x0,
args=(ansatz_isa, Hamiltonian_isa, estimator),
method="cobyla",
options={"maxiter": 10, "tol": 0.01},
)
batch.close()
Iters. done: 1 [Current cost: -0.3361517318448143]
Iters. done: 2 [Current cost: -0.4682546422099432]
Iters. done: 3 [Current cost: -0.38985802144149584]
Iters. done: 4 [Current cost: -0.38319217316749354]
Iters. done: 5 [Current cost: -0.4628720756579032]
Iters. done: 6 [Current cost: -0.4683301936226905]
Iters. done: 7 [Current cost: -0.45480498699294747]
Iters. done: 8 [Current cost: -0.4690533242050814]
Iters. done: 9 [Current cost: -0.465867415110354]
Iters. done: 10 [Current cost: -0.4606882723137227]
h_vqe = res.fun
print("The reference ground state energy is ", min(eigenvalues))
print("The computed ground state energy is ", h_vqe)
The reference ground state energy is (-0.471+0j)
The computed ground state energy is -0.4690533242050814
Félicitations ! Tu viens de terminer avec succès ta première expérience de chimie quantique. On observe une différence entre l'énergie exacte de l'état fondamental de l'Hamiltonien et la nôtre, mais comme nous avons utilisé une technique d'atténuation d'erreurs par défaut (qui corrige les erreurs de lecture), la différence est minime. C'est un très bon début !
Note : Tu peux obtenir un meilleur résultat en définissant un niveau d'atténuation d'erreurs avec resilience_level. La valeur par défaut est 1, et si tu définis une valeur plus élevée, cela utilisera plus de temps QPU mais pourrait retourner un meilleur résultat.
Étape 4 : Post-traitement
Il est temps d'observer comment notre optimiseur classique a travaillé. Exécute la cellule ci-dessous et observe le schéma de convergence.
fig, ax = plt.subplots()
x = np.linspace(0, 10, 10)
# Define the constant function
y_constant = np.full_like(x, h)
ax.plot(
range(cost_history_dict["iters"]), cost_history_dict["cost_history"], label="VQE"
)
ax.set_xlabel("Iterations")
ax.set_ylabel("Cost (Hartree)")
ax.plot(y_constant, label="Target")
plt.legend()
plt.draw()

Nous avons démarré avec une valeur initiale assez bonne, si bien que nous avons obtenu une bonne valeur finale en seulement 10 étapes. Tu peux observer des pics importants et d'autres plus petits — c'est le comportement typique de l'optimiseur COBYLA : il explore l'espace comme s'il ne pouvait pas voir le paysage, et ajuste la taille des pas à chaque mesure.
Vérifie ta compréhension
Quelle est ton observation ? Quelle partie du processus ci-dessus peut être améliorée pour obtenir des résultats plus proches des valeurs théoriques, ou plus proches de l'énergie précise de l'état fondamental de l'Hamiltonien ? Quels sont les éléments à prendre en compte ?
Answer
La première chose à considérer est le changement de l'ensemble de bases utilisé dans le calcul de l'Hamiltonien des molécules. Comme mentionné précédemment, l'énergie de l'état fondamental de l'atome H est -0,5 Hartree, valeur bien connue, et la base STO-6G que nous avons choisie n'est pas suffisante pour dériver cette valeur avec précision.
Choisir un type de base plus complexe augmente le nombre de qubits utilisés par l'Hamiltonien ; il faut donc sélectionner un ansatz plus complexe et mieux adapté aux problèmes de chimie.
La prochaine chose à optimiser est la gestion du bruit dans le QPU. Des techniques d'atténuation d'erreurs plus avancées donnent de meilleurs résultats, mais peuvent prendre plus de temps. Pense également à l'impact du shot_number sur les résultats.
Enfin, de meilleures performances de convergence peuvent également être obtenues en essayant différents optimiseurs.
Calculer l'énergie de l'état fondamental de la molécule d'hydrogène avec VQE
Maintenant que nous avons examiné le processus global de VQE à l'aide d'atomes , nous allons calculer plus rapidement l'énergie de l'état fondamental de la molécule .
Étape 1 : Mapper le problème sur des circuits quantiques et des opérateurs
Nous te fournissons ici un Hamiltonien à un qubit utilisant la base STO-6G et la transformation de Jordan-Wigner, avec réduction du nombre de qubits par exploitation d'une symétrie du Hamiltonien. Note que nous avons utilisé une distance atomique de 0.735 entre les deux atomes d'hydrogène.
Contrairement au calcul d'un atome d'hydrogène isolé (), pour calculer l'état fondamental d'une molécule d'hydrogène (), nous devons également tenir compte de la force répulsive exercée entre les noyaux des deux atomes d'hydrogène, en plus de l'énergie associée aux orbitales électroniques. Dans cette étape, nous donnerons cette valeur comme une constante, et nous calculerons effectivement cette valeur dans le problème de vérification.
h2_hamiltonian = SparsePauliOp.from_list(
[("I", -1.04886087), ("Z", -0.7967368), ("X", 0.18121804)]
)
# exact ground state energy of hamiltonian
nuclear_repulsion = 0.71997
A = np.array(h2_hamiltonian)
eigenvalues, eigenvectors = np.linalg.eig(A)
print("Electronic ground state energy (Hartree): ", min(eigenvalues).real)
print("Nuclear repulsion energy (Hartree): ", nuclear_repulsion)
print(
"Total ground state energy (Hartree): ", min(eigenvalues).real + nuclear_repulsion
)
h2 = min(eigenvalues).real + nuclear_repulsion
Electronic ground state energy (Hartree): -1.8659468547627318
Nuclear repulsion energy (Hartree): 0.71997
Total ground state energy (Hartree): -1.1459768547627318
Étape 2 : Optimiser pour le matériel cible
Étant donné que le nombre de qubits utilisés par le VQE précédent et le Hamiltonien est identique à celui du backend destiné à l'exécution, nous réutiliserons l'ansatz existant et sa forme optimisée.
h2_hamiltonian_isa = h2_hamiltonian.apply_layout(layout=ansatz_isa.layout)
Étape 3 : Exécuter sur le matériel cible
Il est maintenant temps d'effectuer les calculs sur un vrai QPU. Presque tout est identique, mais nous utiliserons un point initial approprié pour ce Hamiltonien. De plus, dans la partie itérative, certains réglages de l'Estimator, qui sert à calculer les valeurs d'espérance du Hamiltonien pour l'ansatz sur le QPU, seront légèrement différents des calculs précédents. Nous reviendrons sur ce changement dans une question de vérification.
x0 = [2, 0, 0]
# QPU time 4min for ibm_brisbane
batch = Batch(backend=backend)
cost_history_dict = {
"prev_vector": None,
"iters": 0,
"cost_history": [],
}
estimator = Estimator(mode=batch)
estimator.options.default_shots = 10000
res = minimize(
cost_func,
x0,
args=(ansatz_isa, h2_hamiltonian_isa, estimator),
method="cobyla",
options={"maxiter": 15},
)
batch.close()
Iters. done: 1 [Current cost: -0.710621837568328]
Iters. done: 2 [Current cost: -0.2603208441168329]
Iters. done: 3 [Current cost: -0.25548711201326424]
Iters. done: 4 [Current cost: -0.581129450619904]
Iters. done: 5 [Current cost: -1.722920997605439]
Iters. done: 6 [Current cost: -1.6633324849371915]
Iters. done: 7 [Current cost: -1.8066989598929164]
Iters. done: 8 [Current cost: -1.8051093803839542]
Iters. done: 9 [Current cost: -1.802692217571555]
Iters. done: 10 [Current cost: -1.8233585485263144]
Iters. done: 11 [Current cost: -1.6904116652617205]
Iters. done: 12 [Current cost: -1.8245120321245392]
Iters. done: 13 [Current cost: -1.6837021361383608]
Iters. done: 14 [Current cost: -1.8166632606115467]
Iters. done: 15 [Current cost: -1.863446212658907]
h2_vqe = res.fun + nuclear_repulsion
print(
"The reference ground state energy is ", min(eigenvalues).real + nuclear_repulsion
)
print("The computed ground state energy is ", h2_vqe)
The reference ground state energy is -1.1459768547627318
The computed ground state energy is -1.143476212658907
Bien que VQE fournisse théoriquement une borne supérieure à la vraie énergie de l'état fondamental, les implémentations concrètes sur du matériel quantique réel ou simulé avec du bruit, ainsi que les approximations effectuées lors de la préparation du Hamiltonien (comme les bases ou la réduction de qubits), peuvent introduire des erreurs qui conduisent parfois à une énergie mesurée légèrement inférieure à la valeur théorique exacte ou à une référence numérique précise. Malgré quelques erreurs, les résultats semblent satisfaisants, notamment compte tenu du faible nombre d'étapes. Examinons maintenant comment l'optimiseur a fonctionné pour terminer ce calcul VQE.
Étape 4 : Post-traitement
fig, ax = plt.subplots()
x = np.linspace(0, 5, 15)
# Define the constant function
y_constant = np.full_like(x, min(eigenvalues))
ax.plot(
range(cost_history_dict["iters"]), cost_history_dict["cost_history"], label="VQE"
)
ax.set_xlabel("Iterations")
ax.set_ylabel("Cost (Hartree)")
ax.plot(y_constant, label="Target")
plt.legend()
plt.draw()

Vérifie ta compréhension
Calculons l'énergie de répulsion nucléaire de la molécule , que nous avons incluse comme valeur constante (0,71997 Hartree).

Utilise la loi de Coulomb et les unités atomiques pour obtenir la valeur en Hartree.
Answer
Étant donné que les deux noyaux d'hydrogène sont chargés positivement, ils se repoussent mutuellement en raison de la force électrostatique. Cette répulsion est décrite par la loi de Coulomb :
où est la charge du proton, est la permittivité du vide, et est la distance entre les deux noyaux, mesurée en mètres ou en rayons de Bohr en unités de joules (J).
Pour calculer cette énergie en Hartrees, nous devons convertir l'équation ci-dessus dans le système d'unités atomiques (UA). En UA, , et le rayon de Bohr () vaut 1, devenant ainsi l'échelle de longueur fondamentale en UA. Grâce à ces simplifications, la loi de Coulomb se réduit à :
où doit être exprimé en rayons de Bohr ().
Pour convertir la séparation nucléaire donnée en en , on utilise la relation de conversion suivante :
donc devient .
Ainsi, l'énergie de répulsion nucléaire de la molécule considérée est
Calculer l'énergie de réaction de
Utilisons maintenant ce que nous avons obtenu ! Tu as utilisé VQE, un solveur quantique variationnel des valeurs propres, pour calculer l'énergie de l'état fondamental de l'atome et de la molécule . Il reste à utiliser ces valeurs calculées pour obtenir l'énergie de réaction du processus .
L'énergie de réaction est le changement d'énergie qui se produit lorsque des substances réagissent pour former de nouvelles substances. Imagine que tu construis quelque chose : parfois tu dois y apporter de l'énergie (comme empiler des blocs), et parfois de l'énergie est libérée (comme une balle qui dévale une pente). En chimie, les réactions absorbent de l'énergie (endothermiques) ou en libèrent (exothermiques).
L'énergie de réaction du processus peut être calculée grâce à la formule suivante :
En exécutant la cellule ci-dessous, visualisons cela graphiquement. Nous utiliserons ici la valeur exacte de l'état fondamental de chaque Hamiltonien, et nous comparerons l'énergie de réaction de la solution exacte avec les résultats de VQE.
# Theoretical values
E_H_theo = h.real
E_H2_theo = h2
# Experimental values
E_H_exp = h_vqe
E_H2_exp = h2_vqe
# Calculate reaction energies
E_reaction_theo = E_H2_theo - (2 * E_H_theo)
E_reaction_exp = E_H2_exp - (2 * E_H_exp)
# Set up the plot
fig, ax = plt.subplots(figsize=(8, 6))
ax.set_xlim(0, 3)
ax.set_ylim(-1.16, -0.93) # Adjust y-axis range to highlight differences
ax.set_xticks([])
ax.set_ylabel("Energy (Hartree)")
ax.set_title("H + H → H₂ Reaction Energy Diagram")
# Plot theoretical energy levels
ax.hlines(
y=2 * E_H_theo, xmin=0.5, xmax=1.3, linewidth=2, color="r", label="2H (Exact)"
)
ax.hlines(y=E_H2_theo, xmin=1.3, xmax=2, linewidth=2, color="b", label="H₂ (Exact)")
# Plot experimental energy levels
ax.hlines(
y=2 * E_H_exp,
xmin=0.5,
xmax=1.5,
linewidth=2,
color="r",
linestyle="dashed",
label="2H (VQE)",
)
ax.hlines(
y=E_H2_exp,
xmin=1.5,
xmax=2.5,
linewidth=2,
color="b",
linestyle="dashed",
label="H₂ (VQE)",
)
# Add labels
ax.text(
1,
2 * E_H_theo,
f"2H: {2*E_H_theo:.4f}",
verticalalignment="top",
horizontalalignment="left",
)
ax.text(
2,
E_H2_theo,
f"H₂: {E_H2_theo:.4f}",
verticalalignment="top",
horizontalalignment="left",
)
ax.text(
1,
2 * E_H_exp,
f"2H_VQE: {2*E_H_exp:.4f}",
verticalalignment="bottom",
horizontalalignment="right",
)
ax.text(
2,
E_H2_exp,
f"H₂_VQE: {E_H2_exp:.4f}",
verticalalignment="bottom",
horizontalalignment="right",
)
# Add arrows for reaction energy with ΔE label in the middle
mid_y_theo = (2 * E_H_theo + E_H2_theo) / 2
mid_y_exp = (2 * E_H_exp + E_H2_exp) / 2
ax.annotate(
"",
xy=(1.3, E_H2_theo),
xytext=(1.3, 2 * E_H_theo),
arrowprops=dict(arrowstyle="<->", color="g"),
)
ax.text(
1.35, mid_y_theo, f"ΔE: {E_reaction_theo:.4f}", color="g", verticalalignment="top"
)
ax.annotate(
"",
xy=(1.5, E_H2_exp),
xytext=(1.5, 2 * E_H_exp),
arrowprops=dict(arrowstyle="<->", color="g", linestyle="dashed"),
)
ax.text(
1.55,
mid_y_exp,
f"ΔE_VQE: {E_reaction_exp:.4f}",
color="g",
verticalalignment="center",
)
# Add legend
ax.legend()
plt.show()

Comme le montre la figure, bien qu'il y ait quelques erreurs, l'énergie exacte de l'état fondamental des Hamiltoniens et l'énergie de réaction calculée à partir des résultats de VQE sont similaires, proches de -0,2 Hartree.
Il convient de noter ici que l'énergie de réaction de ce processus est négative, ce qui signifie que de l'énergie est libérée au cours du processus, et que la molécule résultante possède une énergie inférieure à celle de deux atomes séparés.
- Conclusion
Faisons le point sur ce que nous avons appris jusqu'ici.
Nous avons d'abord examiné deux techniques d'approximation importantes nécessaires pour résoudre des problèmes de chimie quantique : le principe variationnel et le choix des bases, qui sont tous deux fondamentaux pour VQE. Nous avons exploré le principe variationnel à la main, en calculant l'énergie de l'état fondamental de l'oscillateur harmonique simple.
Ensuite, nous avons exploré VQE, un algorithme largement utilisé pour calculer l'énergie de l'état fondamental d'un système quantique. Nous avons exécuté du code pour calculer les énergies de l'état fondamental de l'atome d'hydrogène () et de la molécule d'hydrogène (). En particulier, nous avons appris qu'il est nécessaire d'obtenir le Hamiltonien moléculaire approprié pour le système et de le transformer en une forme exécutable sur un ordinateur quantique. Nous avons également vu que l'ansatz, un circuit quantique paramétré, est nécessaire pour préparer des états quantiques d'essai dans VQE, et nous avons discuté de l'importance du choix d'une structure d'ansatz appropriée. Nous avons également appris que VQE repose sur un processus d'optimisation itératif utilisant un ordinateur classique, guidant le circuit quantique vers l'état d'énergie la plus basse, et nous avons observé comment le processus converge.
Enfin, nous avons utilisé les énergies de l'état fondamental de et obtenues via VQE pour calculer l'énergie de réaction du processus .
VQE est un algorithme quantique puissant à court terme, mais il est important d'être conscient de ses limites. Les performances de VQE dépendent fortement du choix de l'ansatz — trouver un ansatz efficacement préparable qui peut représenter fidèlement le vrai état fondamental devient difficile pour des molécules plus grandes et plus complexes. De plus, le matériel quantique actuel est sensible au bruit, ce qui peut affecter la précision des résultats de VQE, en particulier pour des circuits plus profonds ou un plus grand nombre de qubits. Malgré ces défis, VQE reste un algorithme fondateur, et la recherche en cours explore des méthodes variationnelles plus sophistiquées et des techniques de mitigation des erreurs pour repousser les limites de ce qui est possible en chimie quantique sur des ordinateurs quantiques à court terme. Par exemple, des algorithmes comme la Diagonalisation Quantique par Échantillonnage (SQD, Sample-based Quantum Diagonalization) sont en cours de développement ; ils exploitent des échantillons obtenus à partir de circuits quantiques combinés à une diagonalisation classique dans un sous-espace pour améliorer l'estimation de l'énergie et pallier certaines des limitations de VQE, notamment en ce qui concerne l'efficacité des mesures et la robustesse au bruit.
Révision et questions
Concepts clés :
- L'algorithme quantique variationnel est un paradigme de calcul dans lequel un ordinateur classique et un ordinateur quantique collaborent pour résoudre un problème.
- Dans VQE, on commence avec un Hamiltonien de notre système et on le mappe sur des qubits pour l'exécuter sur l'ordinateur quantique. On sélectionne un circuit quantique paramétré, un ansatz, et on effectue des mesures répétées en faisant varier les paramètres de l'ansatz, jusqu'à atteindre la valeur d'énergie la plus basse. La recherche dans l'espace des paramètres est effectuée à l'aide d'un optimiseur classique. Pour obtenir de bons résultats, il est nécessaire de choisir un bon ansatz et un optimiseur approprié.
- L'énergie de réaction est la variation totale d'énergie lors d'une réaction chimique, déterminée par la différence entre l'énergie des réactifs et celle des produits.
Vrai/Faux
- Le principe variationnel stipule que la valeur d'espérance de l'énergie pour toute fonction d'onde d'essai est toujours supérieure ou égale à la vraie énergie de l'état fondamental.
- Une base est un ensemble de fonctions utilisées pour approximer des fonctions d'onde quantiques.
- VQE est un algorithme quantique permettant de résoudre exactement l'équation de Schrödinger pour un Hamiltonien donné.
- Dans VQE, un circuit quantique paramétré (un ansatz) est utilisé pour préparer des fonctions d'onde d'essai.
- Le choix de l'optimiseur dans VQE (par exemple, COBYLA, SPSA ou ADAM) n'a pas d'impact sur la qualité du résultat.
- L'
Estimatorde Qiskit est utilisé pour calculer directement les valeurs d'espérance des Hamiltoniens dans VQE.
Questions à choix multiples :
- Quel est le rôle du Hamiltonien dans VQE ?
- A) Générer des états quantiques aléatoires
- B) Déterminer l'énergie des états quantiques
- C) Optimiser les circuits quantiques
- D) Créer de l'intrication
- Quel est l'objectif principal de l'algorithme VQE ?
- A) Trouver l'énergie de l'état fondamental d'un Hamiltonien
- B) Créer de l'intrication entre des qubits
- C) Effectuer la recherche de Grover
- D) Casser le chiffrement RSA
- Combien d'états quantiques sont générés dans ce notebook pour comparer l'ansatz ?
- A) 100
- B) 1 000
- C) 5 000
- D) 10 000
- Pourquoi un optimiseur classique est-il nécessaire dans VQE ?
- A) Pour effectuer des mesures quantiques
- B) Mettre à jour les paramètres de l'ansatz pour minimiser l'énergie
- C) Pour intricher des qubits
- D) Pour générer de l'aléatoire quantique
- Pourquoi l'ansatz est-il conçu pour être paramétré ?
- A) Pour permettre la préparation d'états quantiques
- B) Pour permettre d'explorer un large espace d'états quantiques
- C) Pour réduire la complexité du circuit
- D) Pour mesurer directement les valeurs propres
- Laquelle des affirmations suivantes est la plus correcte concernant le choix d'un bon ansatz ?
- A) Un ansatz doit produire des états uniformément distribués sur la sphère de Bloch, sinon il échouera.
- B) Un ansatz doit être adapté à ton système pour s'assurer qu'il peut générer des états proches de l'état fondamental.
- C) Un ansatz doit produire des états aléatoires à l'aide de ses paramètres variationnels.
- D) Un meilleur ansatz a toujours plus de paramètres variationnels.
(Optionnel) Annexe : Surcharge de l'optimiseur selon la complexité de l'ansatz
VQE fait face à plusieurs défis bien connus[ref 6], et les suivants sont liés à ce que nous avons appris ci-dessus.
- Défis liés au choix de l'ansatz
Il existe un défi inhérent dans la sélection du bon ansatz variationnel. Les ansätze inspirés de la chimie (comme UCCSD) offrent une précision physique mais nécessitent des circuits profonds, tandis que les ansätze efficaces pour le matériel ont des circuits plus courts mais peuvent manquer d'interprétabilité physique. De plus, de nombreux ansätze introduisent un excès de paramètres variationnels qui contribuent peu à l'amélioration de la précision mais augmentent considérablement la difficulté de l'optimisation.
- Difficultés d'optimisation
Le paysage d'optimisation du VQE peut comporter des régions où les gradients s'annulent de façon exponentielle (plateaux arides, ou barren plateaus), ce qui rend difficile pour les optimiseurs classiques de mettre à jour efficacement les paramètres variationnels. Pour y remédier, les chercheurs ont essayé différents types d'optimiseurs — basés sur le gradient et sans gradient — mais les deux font face à des défis. Les optimiseurs basés sur le gradient souffrent des plateaux arides, tandis que les méthodes sans gradient nécessitent un grand nombre d'évaluations de la fonction.
- Surcharge de l'optimiseur
Un autre défi bien connu est la surcharge de l'optimiseur, liée à l'échelle du problème. Les circuits quantiques requis par le VQE croissent en profondeur et en complexité à mesure que la taille du problème augmente ; cela accroît généralement aussi le nombre de paramètres à optimiser. Le processus d'optimisation devient intraitable lorsque le nombre de paramètres augmente, entraînant une convergence lente et des difficultés à trouver la solution optimale.
Nous allons maintenant examiner ces défis en utilisant le VQE pour une molécule , avec deux types différents d'ansätze.
(Remarque : cela peut nécessiter plus de temps QPU, alors n'hésite pas à utiliser un simulateur si tu n'as pas suffisamment de temps.)
from qiskit.circuit import ParameterVector
num_iter = 4
alpha = ParameterVector("alpha", 3)
beta = ParameterVector("beta", 3 * num_iter)
# step1: Map problem to quantum circuits and operators
hamiltonian = SparsePauliOp.from_list(
[("I", -1.04886087), ("Z", -0.7967368), ("X", 0.18121804)]
)
ansatz_1 = ansatz3
ansatz_2 = QuantumCircuit(1)
for i in range(num_iter):
ansatz_2.rx(beta[i * 3 + 0], 0)
ansatz_2.rz(beta[i * 3 + 1], 0)
ansatz_2.rx(beta[i * 3 + 2], 0)
ansatz_1.draw("mpl")
ansatz_2.draw("mpl")

# Step 2: Optimize for target hardware
target = backend.target
pm = generate_preset_pass_manager(target=target, optimization_level=3)
ansatz_isa_1 = pm.run(ansatz_1)
ansatz_isa_2 = pm.run(ansatz_2)
hamiltonian_isa_1 = hamiltonian.apply_layout(layout=ansatz_isa_1.layout)
hamiltonian_isa_2 = hamiltonian.apply_layout(layout=ansatz_isa_2.layout)
Exécutons maintenant un VQE avec un point de départ composé entièrement de uns, avec un maximum de 20 étapes, et comparons la convergence des deux exécutions.
# QPU time 3m 40s for ibm_brisbane
# Step 3: Execute on target hardware
from scipy.optimize import minimize
x0 = np.ones(ansatz_1.num_parameters)
batch = Batch(backend=backend)
cost_history_dict = {
"prev_vector": None,
"iters": 0,
"cost_history": [],
}
estimator = Estimator(mode=batch)
estimator.options.default_shots = 2048
res = minimize(
cost_func,
x0,
args=(ansatz_isa_1, hamiltonian_isa_1, estimator),
method="cobyla",
options={"maxiter": 20},
)
batch.close()
Iters. done: 1 [Current cost: -0.8782202668652658]
Iters. done: 2 [Current cost: -0.43473160695469165]
Iters. done: 3 [Current cost: -0.4076372093159749]
Iters. done: 4 [Current cost: -1.3587839859772106]
Iters. done: 5 [Current cost: -1.774529906754082]
Iters. done: 6 [Current cost: -1.541934983115727]
Iters. done: 7 [Current cost: -1.2732403113465345]
Iters. done: 8 [Current cost: -1.820842221085785]
Iters. done: 9 [Current cost: -1.8065762857059005]
Iters. done: 10 [Current cost: -1.8126394095981146]
Iters. done: 11 [Current cost: -1.8205831886180421]
Iters. done: 12 [Current cost: -1.8086715778994924]
Iters. done: 13 [Current cost: -1.8307676638629322]
Iters. done: 14 [Current cost: -1.8177328827556327]
Iters. done: 15 [Current cost: -1.8179426218088064]
Iters. done: 16 [Current cost: -1.8109239667991088]
Iters. done: 17 [Current cost: -1.824271872489647]
Iters. done: 18 [Current cost: -1.813167587671394]
Iters. done: 19 [Current cost: -1.824647343397313]
Iters. done: 20 [Current cost: -1.8219785311686143]
# Save Cost_history as a new list
ansatz_1_history = cost_history_dict["cost_history"]
# QPU time 3m 40s for ibm_brisbane
x0 = np.ones(ansatz_2.num_parameters)
batch = Batch(backend=backend)
cost_history_dict = {
"prev_vector": None,
"iters": 0,
"cost_history": [],
}
estimator = Estimator(mode=batch)
estimator.options.default_shots = 2048
res = minimize(
cost_func,
x0,
args=(ansatz_isa_2, hamiltonian_isa_2, estimator),
method="cobyla",
options={"maxiter": 20},
)
batch.close()
Iters. done: 1 [Current cost: -0.738191173881188]
Iters. done: 2 [Current cost: -0.42636037194506304]
Iters. done: 3 [Current cost: -1.3503788613797374]
Iters. done: 4 [Current cost: -0.9109204349776897]
Iters. done: 5 [Current cost: -0.9060873157510835]
Iters. done: 6 [Current cost: -0.7735065414083984]
Iters. done: 7 [Current cost: -1.586889197437709]
Iters. done: 8 [Current cost: -1.659215191584943]
Iters. done: 9 [Current cost: -1.245445981794618]
Iters. done: 10 [Current cost: -1.1608385766138023]
Iters. done: 11 [Current cost: -1.1551733876027737]
Iters. done: 12 [Current cost: -1.8143337768286332]
Iters. done: 13 [Current cost: -1.2510951563756598]
Iters. done: 14 [Current cost: -1.6918311531865413]
Iters. done: 15 [Current cost: -1.8163783305531838]
Iters. done: 16 [Current cost: -1.8434877732947152]
Iters. done: 17 [Current cost: -1.8461898233304472]
Iters. done: 18 [Current cost: -1.0346471214915485]
Iters. done: 19 [Current cost: -1.8322518854150687]
Iters. done: 20 [Current cost: -1.717144678705999]
ansatz_2_history = cost_history_dict["cost_history"]
fig, ax = plt.subplots()
# Define the constant function)
ax.plot(
range(cost_history_dict["iters"]),
ansatz_1_history,
label="Ansatz with 3 parameters",
)
ax.plot(
range(cost_history_dict["iters"]),
ansatz_2_history,
label="Ansatz with 12 parameters",
)
ax.set_xlabel("Iterations")
ax.set_ylabel("Cost (Hartree)")
plt.legend()
plt.draw()

Le graphique ci-dessus montre clairement que le processus d'optimisation de l'ansatz avec davantage de variables met plus de temps à atteindre une convergence stable.
Plutôt que de s'appuyer sur de simples circuits à un seul qubit et un ansatz direct, la complexité de l'optimisation augmente lorsque des circuits quantiques plus grands et des ansätze à structure plus complexe sont nécessaires. Cela met en évidence un défi bien connu des VQE : la surcharge de l'optimiseur.
Les chercheurs continuent de développer diverses méthodologies avancées permettant d'utiliser les ordinateurs quantiques pour les problèmes de chimie. Tu peux accéder à une variété de ressources pédagogiques sur IBM Quantum Learning.
Références
- [ref 1] Richard P. Feynman, Simulating Physics with Computers, International Journal of Theoretical Physics, 1982.
- [ref 2] Marov, M.Y. (2015). The Structure of the Universe. In: The Fundamentals of Modern Astrophysics. Springer, New York, NY.
- [ref 3] How to solve difficult chemical engineering problems with quantum computing, IBM Research Blog, 2023.
- [ref 4] Y. Cao, J. Romero and A. Aspuru-Guzik, "Potential of quantum computing for drug discovery," in IBM Journal of Research and Development, vol. 62, no. 6, pp. 6:1-6:20, 1 Nov.-Dec. 2018
- [ref 5] Present State of Molecular Structure Calculation, REv. Mod. Phys. 32, 170, 1960
- [ref 6] Fedorov, D.A., Peng, B., Govind, N. et al. VQE method: a short survey and recent developments. Mater Theory 6, 2 (2022)