HI-VQE Chemistry - Une fonction Qiskit par Qunova Computing
# Added by doQumentation — required packages for this notebook
!pip install -q qiskit-ibm-catalog qiskit-ibm-runtime
# This cell is hidden from users
from qiskit_ibm_runtime import QiskitRuntimeService
service = QiskitRuntimeService()
instance = service.active_account()["instance"]
backend_name = service.least_busy(operational=True, min_num_qubits=16).name
Voir la référence API
Les fonctions Qiskit sont une fonctionnalité expérimentale disponible uniquement pour les utilisateurs des plans IBM Quantum® Premium, Flex et On-Prem (via l'API IBM Quantum Platform). Elles sont en version préliminaire et susceptibles d'évoluer.
Package versions
Le code de cette page a été développé avec les dépendances suivantes. Nous recommandons d'utiliser ces versions ou des versions plus récentes.
qiskit-ibm-runtime~=0.45.0
Vue d'ensemble
En chimie quantique, le problème de la structure électronique consiste à trouver les solutions à l'équation de Schrödinger électronique — les fonctions d'onde quantiques décrivant le comportement des électrons du système. Ces fonctions d'onde sont des vecteurs d'amplitudes complexes, chaque amplitude correspondant à la contribution d'une configuration électronique possible.
L'état fondamental est la fonction d'onde de plus basse énergie du système et revêt une importance particulière dans l'étude des systèmes moléculaires. L'approche la plus précise pour calculer l'état fondamental prend en compte toutes les configurations électroniques possibles, mais cela devient insoluble pour les systèmes plus grands, car le nombre de configurations croît de façon exponentielle avec la taille du système.
Le Handover Iterative Variational Quantum Eigensolver (HI-VQE) est une méthode hybride quantique-classique innovante pour estimer avec précision l'état fondamental de systèmes moléculaires. Il intègre le matériel quantique et le calcul classique : les processeurs quantiques explorent efficacement les configurations électroniques candidates, tandis que la fonction d'onde résultante est calculée sur des ordinateurs classiques. En générant des fonctions d'onde compactes mais chimiquement précises, HI-VQE favorise la recherche et la découverte en chimie quantique et en science des matériaux.

HI-VQE réduit la complexité calculatoire du problème de structure électronique en estimant l'état fondamental avec une grande précision. Il se concentre sur un sous-ensemble soigneusement sélectionné des configurations électroniques les plus pertinentes, optimisant à la fois la précision et l'efficacité.
En combinant les atouts des ordinateurs classiques et quantiques, HI-VQE affine et améliore itérativement l'estimation courante de la fonction d'onde. Ses techniques uniques de construction de sous-espace rendent la sélection des configurations plus efficace, offrant aux utilisateurs un meilleur contrôle calculatoire et une précision accrue dans les simulations de chimie quantique.
Pour en savoir plus sur l'algorithme, tu peux lire l'article de recherche associé.
Description
Le nombre de configurations électroniques d'un système moléculaire croît de façon exponentielle avec la taille du système. Cependant, pour certains états électroniques, comme l'état fondamental, seule une petite fraction des configurations contribue de manière significative à l'énergie de l'état. Les méthodes d'interaction de configuration sélectionnée (SCI) exploitent cette parcimonie pour réduire les coûts calculatoires en identifiant et en se concentrant sur les configurations les plus pertinentes. Ce sous-ensemble de configurations est appelé sous-espace.
HI-VQE tire parti de l'efficacité inhérente des ordinateurs quantiques pour représenter les systèmes moléculaires et faciliter la recherche de sous-espace. Il intègre des sous-routines classiques et quantiques pour résoudre le problème de structure électronique avec une grande précision. Contrairement aux méthodes SCI quantiques existantes, HI-VQE combine entraînement variationnel, construction itérative de sous-espace et filtrage de configurations par pré-diagonalisation pour améliorer l'efficacité en réduisant le nombre de mesures quantiques, d'itérations et les coûts de diagonalisation classique. HI-VQE peut donc être appliqué à des systèmes moléculaires plus grands nécessitant plus de qubits, et réduit le coût de résolution d'un problème d'une taille donnée au même degré de précision.
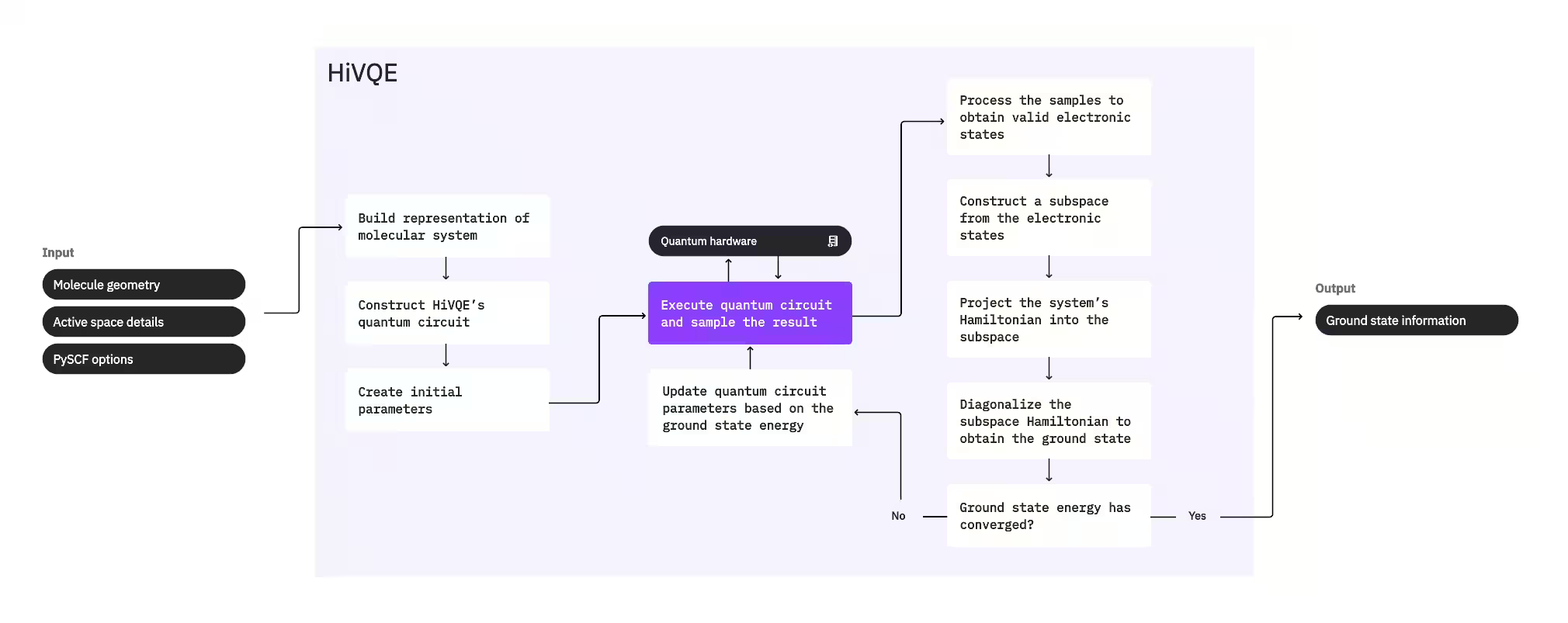
Pour calculer l'état fondamental d'un système, HI-VQE utilise d'abord le package de chimie classique PySCF pour générer une représentation moléculaire à partir des entrées fournies par l'utilisateur, telles que la géométrie moléculaire et d'autres informations moléculaires. Il entre ensuite dans une boucle d'optimisation hybride quantique-classique, affinant itérativement un sous-espace pour représenter optimalement l'état fondamental tout en minimisant le nombre de configurations incluses. La boucle se poursuit jusqu'à ce que les critères de convergence — comme la taille du sous-espace ou la stabilité de l'énergie — soient satisfaits, après quoi la fonction d'onde de l'état fondamental calculée et l'énergie sont produites en sortie. Ces résultats peuvent être utilisés pour construire des surfaces d'énergie potentielle précises et effectuer une analyse plus approfondie du système.
La boucle d'optimisation se concentre sur l'ajustement des paramètres d'un circuit quantique pour générer un sous-espace de haute qualité. HI-VQE propose trois options de circuit quantique : excitation_preserving, efficient_su2 et LUCJ. L'optimisation est initialisée près de l'état de référence Hartree-Fock en raison de son adéquation générale. Le circuit est ensuite exécuté sur un dispositif quantique et des configurations sont échantillonnées à partir de l'état quantique résultant avant d'être renvoyées sous forme de chaînes binaires. En raison du bruit des dispositifs quantiques, certaines configurations échantillonnées peuvent être physiquement invalides, ne conservant pas le nombre d'électrons ou le spin. HI-VQE résout ce problème grâce au processus de récupération de configuration du package qiskit-addon-sqd, permettant aux utilisateurs de corriger les configurations invalides ou de les rejeter.
Les configurations valides passent ensuite par une étape de filtrage optionnelle pour éliminer celles dont la contribution est prédite comme minimale. Cela réduit la dimension du sous-espace, diminuant ainsi le coût de l'étape de diagonalisation. Si le filtrage est activé, un Hamiltonien de sous-espace préliminaire est construit à partir des configurations valides et une diagonalisation est effectuée avec des critères d'arrêt très souples. Bien que la précision des amplitudes résultantes pour chaque configuration soit faible, cela est efficace pour prédire quelles configurations exclure du sous-espace lors de cette itération, et ce calcul est rapide.
Les configurations sélectionnées sont ajoutées au sous-espace, et le Hamiltonien du système est projeté dans ce sous-espace. Le sous-espace se met à jour itérativement, conservant les configurations les plus pertinentes d'une itération à l'autre. Cette approche contraste avec les méthodes alternatives car le circuit quantique n'a pas besoin d'approximer l'intégralité de l'état fondamental à chaque étape.
Ensuite, le Hamiltonien de sous-espace est diagonalisé classiquement pour obtenir la valeur propre la plus basse et son vecteur propre correspondant, représentant une approximation de l'état fondamental et de son énergie. À mesure que la qualité du sous-espace s'améliore au fil des itérations, l'état fondamental calculé se rapproche davantage du vrai état fondamental. Une étape de filtrage supplémentaire peut être effectuée à ce stade pour retirer du sous-espace toute configuration ne contribuant pas substantiellement à l'état fondamental calculé. Cette étape garantit que le sous-espace transmis à l'itération suivante est aussi compact que possible. Elle est évaluée sur la base des amplitudes renvoyées par la diagonalisation, qui représentent la contribution d'importance de chaque configuration à l'état fondamental calculé.
Un test de convergence détermine ensuite si un entraînement supplémentaire améliorerait les résultats. Dans l'affirmative, une étape d'expansion classique optionnelle est effectuée, les paramètres du circuit quantique sont mis à jour pour minimiser davantage l'énergie calculée, et le processus recommence. L'étape d'expansion classique génère des configurations supplémentaires pour le sous-espace, venant compléter les configurations échantillonnées depuis le dispositif quantique. Elle identifie d'abord la configuration ayant la plus grande amplitude dans les résultats de diagonalisation, puis génère de nouvelles configurations avec des excitations simples et doubles à partir de la configuration identifiée. Le nombre souhaité de ces configurations est ensuite ajouté au sous-espace.
Une fois qu'il est déterminé que les itérations ont convergé, HI-VQE renvoie l'état fondamental calculé (sous la forme des états dans le sous-espace et leurs amplitudes dans la fonction d'onde de l'état fondamental), son énergie, et une mesure de la variance d'énergie indiquant si l'état calculé forme un état propre du Hamiltonien du système.
Les utilisateurs peuvent choisir le circuit quantique utilisé et le nombre de shots pris pour chaque circuit quantique, ainsi que contrôler la taille du sous-espace ou activer la génération classique de configurations supplémentaires pour compléter les configurations générées quantiquement. Ainsi, les utilisateurs peuvent adapter le comportement de HI-VQE à leurs applications.
Licence
Note que l'utilisation de cette fonction Qiskit est limitée aux problèmes nécessitant au maximum 20 qubits, sauf si une licence accordant une limite plus élevée est obtenue.
Envoie un e-mail à qiskit.support@qunovacomputing.com si tu souhaites te renseigner sur l'obtention d'une licence.
Démarrage
D'abord, demande l'accès à la fonction. Ensuite, authentifie-toi avec ta clé API IBM Quantum® et, en supposant que tu as déjà enregistré ton compte dans ton environnement local, sélectionne la fonction Qiskit comme suit :
import reprlib
from qiskit_ibm_catalog import QiskitFunctionsCatalog
catalog = QiskitFunctionsCatalog(channel="ibm_quantum_platform")
function = catalog.load("qunova/hivqe-chemistry")
Exemple
Le premier exemple montre comment calculer l'énergie de l'état fondamental d'une molécule NH3 à l'aide de l'algorithme HI-VQE.
Définir la géométrie moléculaire et les options
La géométrie moléculaire de NH3 est fournie avec des coordonnées cartésiennes séparées par « ; » pour chaque atome.
# Define the molecule geometry
geometry = """
N -0.85188 -0.02741 0.03141;
H 0.16545 0.00593 -0.01648;
H -1.16348 -0.39357 -0.86702;
H -1.16348 0.94228 0.06281;
"""
Des options supplémentaires peuvent être définies et fournies pour le système moléculaire dans le format de dictionnaire suivant.
# Configure some options for the job.
molecule_options = {"basis": "sto3g"}
hivqe_options = {"shots": 100, "max_iter": 20}
Exécute la fonction avec les entrées de géométrie et d'options.
# Run HI-VQE
job = function.run(
geometry=geometry,
# `backend_name` is the name of a backend with at least 16 qubits,
# for example, "ibm_marrakesh".
backend_name=backend_name,
max_states=2000,
max_expansion_states=10,
molecule_options=molecule_options,
hivqe_options=hivqe_options,
)
Il est conseillé d'afficher l'ID du job de la fonction afin de pouvoir le fournir dans les demandes de support en cas de problème.
print("Job ID:", job.job_id)
Job ID: e5ced6f2-fd1d-4244-a6aa-bd27cfb0cdee
Cet exemple utilise ensuite 16 qubits avec 8 orbitales de base sto3g pour une molécule NH3. Vérifie le statut de ta charge de travail de fonction Qiskit ou récupère les résultats comme suit :
print(job.status())
QUEUED
Une fois le job terminé, les résultats peuvent être obtenus avec l'instance result().
result = job.result()
# Output can be long, so we display a shortened representation
shortened_result = reprlib.repr(result)
print(shortened_result)
{'eigenvector': [0.9824448589364075, 0.009527106392132133, 6.854074372058527e-08, 3.591500190038039e-07, 0.0012975231577544268, 2.310159709002111e-05, ...], 'energy': -55.52108557170985, 'energy_history': [-55.51901898989887, -55.52056881448526, -55.52065046778772, -55.520690696813716, -55.520691108428, -55.520708448092634, ...], 'energy_variance': 3.066239097617371e-10, ...}
Pour accéder à l'énergie de l'état fondamental, utilise la clé "energy". La clé "eigenvector" fournit les coefficients CI avec la notation en chaîne de bits correspondante de la configuration électronique stockée dans "states" des résultats.
fci_energy = -55.521148034704126 # the exact energy using FCI method
hivqe_energy = result["energy"]
print(
f"|Exact Energy - HI-VQE Energy|: "
f"{abs(fci_energy - hivqe_energy) * 1000} mHa"
)
print(f"Sampled Number of States: {len(result['states'])}")
|Exact Energy - HI-VQE Energy|: 0.06246299427914437 mHa
Sampled Number of States: 1936
Performance
Cette section présente les calculs de référence démontrés de HI-VQE avec un cas à 24 qubits pour Li2S, un cas à 40 qubits pour une molécule N2 et un cas à 44 qubits pour un système FeP-NO.
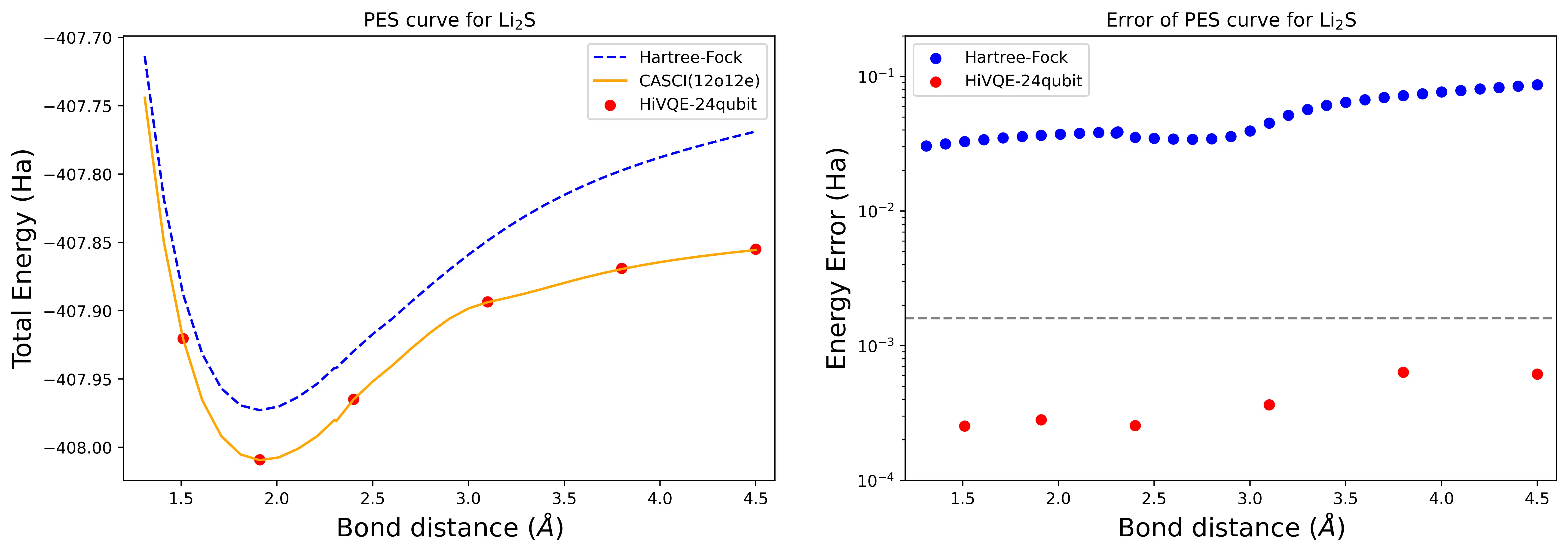
Courbe de surface d'énergie potentielle de dissociation pour une molécule Li2S avec 24 qubits
La courbe PES est montrée avec la référence FCI et l'estimation initiale de RHF, ainsi que l'erreur d'énergie par rapport à la référence FCI.
Les calculs ont été effectués avec les géométries et options suivantes.
# This cell is hidden from users
backend_name = service.least_busy(operational=True, min_num_qubits=38).name
# Define Li2S geometries
Li2S_geoms = {
"Li2S_1.51": "S -1.239044 0.671232 -0.030374;Li -1.506327 0.432403 -1.498949;Li -0.899996 0.973348 1.826768;",
"Li2S_2.40": "S -1.741432 0.680397 0.346702;Li -0.529307 0.488006 -1.729343;Li -1.284307 0.989409 2.177209;",
"Li2S_3.80": "S -2.707255 0.674298 0.909161;Li 0.079218 0.552012 -1.671656;Li -0.927010 0.931502 1.557063;",
}
# Configure some options for the job.
molecule_options = {
"basis": "sto3g",
}
hivqe_options = {
"shots": 100,
"max_iter": 20,
}
results = []
for geom in ["Li2S_1.51", "Li2S_2.40", "Li2S_3.80"]:
# Run HI-VQE
job = function.run(
geometry=Li2S_geoms[geom],
backend_name=backend_name, # can use any device with at least 38 qubits
max_states=2000,
max_expansion_states=10,
molecule_options=molecule_options,
hivqe_options=hivqe_options,
)
results.append(job.result())
Les points rouges représentent les résultats de calcul HI-VQE pour six géométries différentes, et trois géométries correspondant à 1,51, 2,40 et 3,80 angströms sont fournies en entrée dans la cellule ci-dessus.
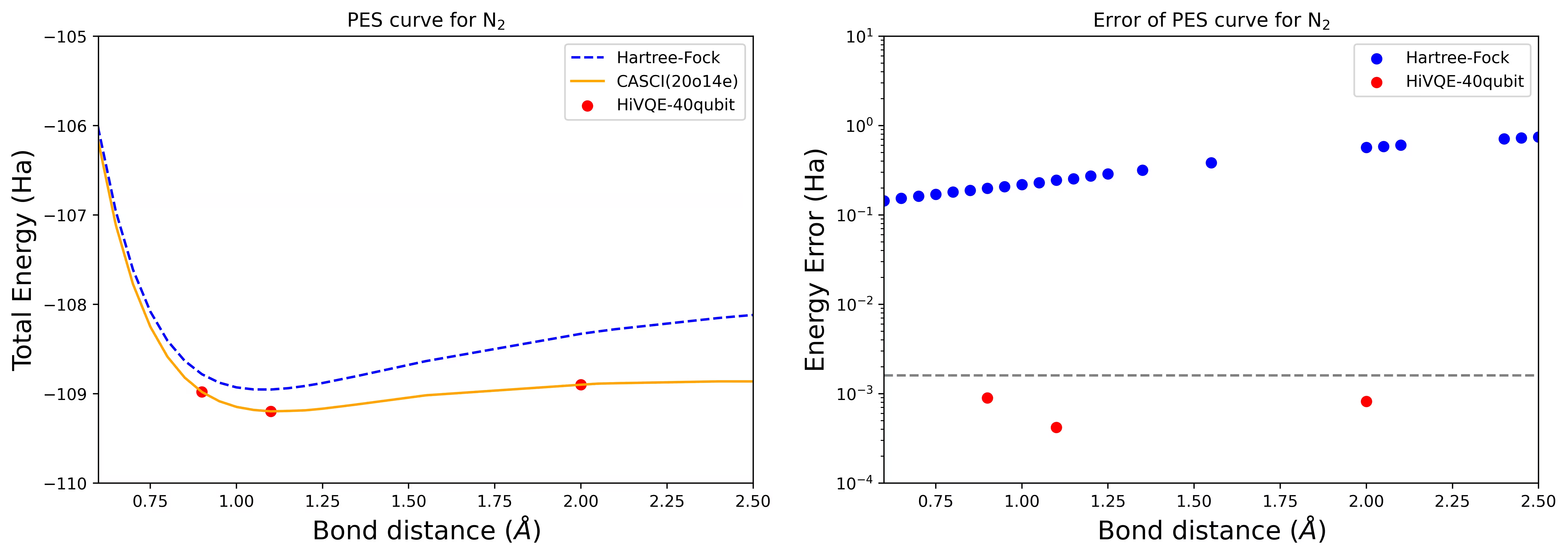
Courbe PES de dissociation pour une molécule N2 avec 40 qubits
La molécule d'azote a été identifiée comme un système multiréférence avec de grandes contributions d'énergie de corrélation au-delà de l'état Hartree-Fock. Nous avons effectué un calcul de référence pour la molécule N2 avec la base cc-pvdz, (20o,14e) en utilisant la sélection d'orbitales actives homo-lumo. Le nombre d'espace actif complet (CAS) pour représenter ce problème est 6 009 350 400. Il n'est pas possible d'obtenir la solution au problème des valeurs propres (pour l'énergie et la structure électronique) avec ce nombre d'états en utilisant un ordinateur de bureau puissant (16 cpu/64 Go). Avec HI-VQE, les utilisateurs peuvent efficacement explorer le sous-espace des états CAS pour trouver des résultats chimiquement précis tout en économisant considérablement les ressources de calcul. Les tracés suivants montrent la courbe PES du calcul HI-VQE à 40 qubits pour la dissociation de la molécule N2.
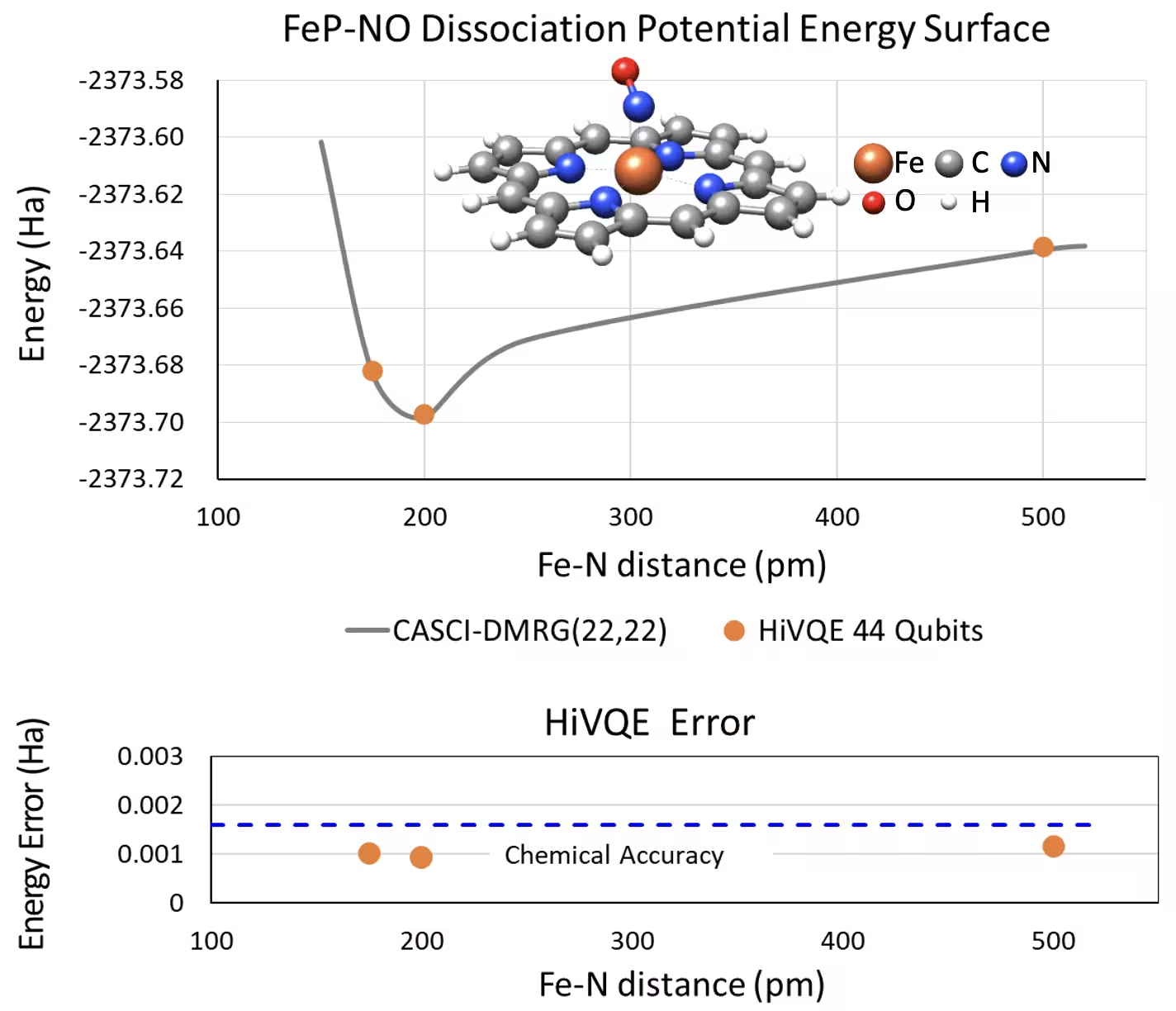
Courbe PES de dissociation pour la porphyrine de fer(II) pentacoordinée avec un système NO à 44 qubits
Un autre système chimique intéressant est un complexe de porphyrine de fer(II) (FeP) avec un ligand monoxyde d'azote (NO) coordiné, qui représente un système métalloporphyrinique biologiquement pertinent jouant un rôle crucial dans divers processus physiologiques. Dans cet exemple, HI-VQE a été utilisé pour estimer la courbe de surface d'énergie potentielle précise de l'interaction intermoléculaire entre FeP et NO (énergie de l'état fondamental pour des géométries séparées différemment). Le système combiné compte 450 orbitales et 202 électrons (450o,202e) avec la base 6-31g(d) au total. La sélection d'orbitales actives homo-lumo a été utilisée pour calculer le cas réduit à partir du cas réel avec (22o,22e). D'après les résultats de référence suivants, nous avons pu atteindre la précision chimique (> 1,6 mHa) avec un calcul de chimie par ordinateur classique de pointe CASCI(DMRG) (22o,22e) de référence.
Points de référence
- La taille exacte de la matrice est le nombre de déterminants pour la solution exacte, comme FCI et CASCI.
- Le calcul HI-VQE échantillonne et calcule le sous-espace de celle-ci (c'est-à-dire la taille de la matrice HI-VQE).
- Le temps total comprend le temps d'exécution QPU et les exécutions de la fonction Qiskit avec CPU.
- La précision est estimée à partir de la différence d'énergie par rapport à la solution exacte.
| Système chimique | Nombre de qubits | Taille exacte de matrice | Taille matrice HI-VQE | E(diff) de l'exact (mHa) | Nombre d'itérations | Temps total | Utilisation du temps QPU |
|---|---|---|---|---|---|---|---|
| (8o,10e) | 16 | 3136 | 1936 | 0,08 | 6 | 37 s | 34 s |
| (10o,10e) | 20 | 63504 | 3969 | 0,60 | 5 | 250 s | 50 s |
| (15o,10e) | 30 | 9018009 | 49729 | 0,90 | 5 | 354 s | 54 s |
| (16o,14e) | 32 | 130873600 | 1798281 | 1,10 | 9 | 6531 s | 121 s |
| (18o,24e) | 36 | 344622096 | 399424 | 0,90 | 24 | 5174 s | 130 s |
| (20o,14e) | 40 | 6009350400 | 9012004 | 1,20 | 21 | 46547 s | 258 s |
Récupérer les messages d'erreur
Si ta charge de travail échoue, le statut sera ERROR et l'appel de job.result() lèvera une exception :
job = function.run(
geometry="invalid-geometry", # This will cause an error
backend_name=backend_name,
max_states=2000,
max_expansion_states=15,
molecule_options=molecule_options,
hivqe_options=hivqe_options,
)
job.result()
job.status()
'ERROR'
Obtenir de l'aide
Tu peux envoyer un e-mail à qiskit.support@qunovacomputing.com pour obtenir de l'aide avec cette fonction.
Si tu souhaites de l'aide pour résoudre une erreur spécifique, fournis l'ID du job de la fonction qui a rencontré l'erreur.
Prochaines étapes
- Demande l'accès à la fonction en remplissant ce formulaire.
- Consulte la référence API pour cette fonction Qiskit.
- Essaie le tutoriel Calculer la courbe PES de dissociation pour FeP-NO avec HI-VQE.
- Consulte Pellow-Jarman, A., et al. (2025). HIVQE : Handover Iterative Variational Quantum Eigensolver for Efficient Quantum Chemistry Calculations. arXiv preprint arXiv:2503.06292.
- Essaie le tutoriel Courbes PES de dissociation avec Qunova HiVQE.