Correspondance
Regarde la vidéo sur la correspondance présentée par Olivia Lanes, ou ouvre la vidéo dans une fenêtre séparée sur YouTube.
Introduction
Dans cette leçon, nous allons nous concentrer sur la première étape — et souvent la plus difficile — de la définition d'un programme quantique : la correspondance d'un problème à exécuter sur un ordinateur quantique. Cette étape couvre la façon dont un utilisateur part d'un problème informatique et le traduit en quelque chose qui peut être résolu sur un ordinateur quantique.
Dans les leçons 2 et 3 de ce cours, nous avons mentionné que l'étape de correspondance est la première des quatre étapes du cadre Qiskit patterns. D'après ces leçons, tu te souviens peut-être que l'objectif de la correspondance est de traduire ou de réécrire un problème informatique sous la forme d'une fonction de coût ou d'une valeur d'espérance que l'on peut évaluer à l'aide d'un ordinateur quantique.
Dans la leçon 3, nous avons discuté d'un exemple concret avec le Max-Cut, un problème computationnellement difficile mais très courant en optimisation combinatoire. Dans cet exemple, nous avons suivi plusieurs étapes pour traduire le problème de graphe initial en un problème pouvant être résolu sur un ordinateur quantique. Nous avons transformé le problème consistant à trouver le nombre maximal de coupes dans le graphe en une fonction de coût, réécrit cette fonction de coût sous forme de Hamiltonien, puis préparé un état quantique d'essai dont l'état fondamental correspondait à la coupe maximale. Enfin, nous avons construit un circuit quantique représentant l'état quantique d'essai qui nous intéressait, puis ajouté les portes spécifiques pour permettre à l'état d'évoluer dans le temps. Cette séquence d'étapes faisait entièrement partie de la correspondance. Bien que les étapes exactes fussent propres au problème Max-Cut, la même procédure générale peut être appliquée à de nombreuses autres applications, comme la chimie quantique et les simulations quantiques.
La correspondance peut être difficile. Il n'existe pas de stratégie universelle pour chaque problème, ce qui peut être intimidant. Dans cette leçon, nous allons examiner quelques considérations générales sur la correspondance, puis plonger dans des exemples représentatifs pour illustrer les différentes façons de mettre un problème en correspondance avec un ordinateur quantique.
Considérations générales
Bien que la stratégie exacte utilisée pour mettre un problème en correspondance avec un ordinateur quantique dépende du problème en question, ces stratégies accomplissent généralement quelques éléments essentiels.
Premièrement, tu pourrais avoir besoin de simplifier le problème pour le rendre faisable. Ce n'est pas propre au quantique — toutes les disciplines scientifiques utilisent des modèles simplifiés pour étudier les phénomènes qui les intéressent, tout en ignorant les détails non pertinents. En physique, il existe une expression célèbre qui pousse ce principe à l'extrême : « suppose une vache sphérique ». Il est souvent trop difficile de décrire un système exactement tel qu'il apparaît, mais on peut plutôt faire des simplifications raisonnables qui peuvent quand même mener à des solutions utiles. Quelques exemples de la façon dont nous pourrions procéder en informatique quantique : limiter la taille ou la profondeur du circuit en choisissant un ansatz adapté au matériel, tronquer des évolutions temporelles complexes, ou négliger les termes du Hamiltonien qui contribuent peu à l'énergie finale d'un état quantique.
Deuxièmement, la correspondance implique d'écrire le problème de manière à ce qu'un ordinateur quantique puisse le comprendre. Cela implique souvent de se poser ces trois questions :
- Que représenteront nos qubits dans notre modèle ?
- Notre problème est-il continu ? Puisque les ordinateurs quantiques sont numériques, si le problème est continu, nous devrons trouver un moyen de le discrétiser.
- Enfin, la topologie du problème correspond-elle à la topologie du matériel ? Si ce n'est pas le cas, nous pourrions avoir besoin d'astuces pour le faire fonctionner.
Abordons la première question, qui est au cœur de nombreuses difficultés liées à la compréhension de la correspondance : que peut représenter un qubit ?
Les qubits peuvent être utilisés pour représenter beaucoup de choses. La première, et peut-être la plus simple, est un nœud sur un graphe. Les graphes sont utilisés pour montrer la connectivité dans de nombreux types différents de problèmes mathématiques, et les nœuds sont un élément fondamental représentant un point ou une entité au sein d'un réseau. Selon ce que représente l'ensemble du réseau, cela pourrait être une ville, une personne, ou un ferromagnet dans un réseau cristallin.
Les qubits peuvent également être utilisés pour représenter des bosons et des fermions, bien que je te mette en garde ici : un seul qubit ne correspond pas exactement à un boson ou à un fermion — c'est un peu plus compliqué que ça, comme nous en discuterons plus loin dans la leçon.
Voici maintenant des exemples un peu plus compliqués. Pour ces modèles, il n'est plus pertinent de parler en termes de qubits individuels ; à la place, nous avons besoin de groupes de qubits pour constituer quelque chose de physique. Par exemple, un groupe de qubits, ici représenté sur une topologie hexagonale lourde, peut être utilisé pour représenter les emplacements géométriques des acides aminés ; des chaînes polymères. Un autre exemple est la simulation de la diffusion des hadrons dans des modèles de physique des hautes énergies, qui peut être réalisée en simulant l'évolution temporelle d'un paquet d'ondes hadronique. Dans ce cas, un registre de qubits peut être utilisé pour encoder différents états d'un champ quantique : l'état du vide de ce champ, ou un paquet d'ondes qui se propage au-dessus de ce vide.
Mais à ce stade, nous avons parlé de manière suffisamment abstraite du défi qui nous attend. Examinons ces exemples en détail.
Exemples de correspondance
Max-Cut
Commençons par notre premier exemple. L'un des problèmes de correspondance les plus directs est celui que nous avons déjà couvert en profondeur : l'exemple du Max-Cut. Dans ce problème, la correspondance était assez simple car un qubit était équivalent à un nœud sur notre graphe.
Rappelle-toi que, pour mettre en correspondance le problème Max-Cut, nous avons exprimé la fonction de coût sous forme de Hamiltonien en utilisant la formulation QUBO. Une fonction de coût Hamiltonienne est une fonction qui encode la solution optimale du problème dans l'état fondamental du Hamiltonien. Pour construire le Hamiltonien de coût, nous avons utilisé la classe SparsePauliOp dans Qiskit pour spécifier la connectivité de notre graphe, et l'étape de correspondance vers les opérateurs quantiques était terminée. Le circuit quantique était simplement l'ansatz QAOA. Pour une remise à niveau, consulte la leçon QAOA à l'échelle utilitaire, où nous détaillons tout cela bien plus en profondeur.
Dans cette leçon, même dans l'exemple à 100 qubits à l'échelle utilitaire, la connectivité du graphe correspondait déjà à la topologie de notre matériel supraconducteur. Nous n'avions donc pas à nous préoccuper de gérer des topologies différentes. Mais ce n'est pas toujours le cas. Si nous avions un graphe plus compliqué ou plus densément connecté que les exemples que nous avons mis en avant jusqu'ici, nous aurions besoin d'implémenter une série de portes SWAP pour modifier la connectivité effective du matériel. Cela est géré à la deuxième étape de Qiskit patterns, la transpilation, mais doit être gardé à l'esprit même dès l'étape de correspondance.
Repliement des protéines
Explorons ensuite un exemple modélisé dans l'article intitulé « Resource-efficient quantum algorithm for protein folding », rédigé par IBM® et des collaborateurs de l'Université de Nouvelle-Galles du Sud.
Un peu de biochimie comme contexte : les protéines sont des macromolécules composées de longues chaînes d'acides aminés. Ces chaînes se replient en structures complexes qui remplissent une grande variété de fonctions biologiques. Déterminer la structure d'une protéine dans l'espace tridimensionnel, et comprendre les relations entre structure et fonction, figurent parmi les problèmes les plus difficiles de la biochimie aujourd'hui. Les protéines se replient en structures utiles grâce aux interactions entre les acides aminés. Au fur et à mesure qu'une structure se tord et se replie, des acides aminés qui sont éloignés les uns des autres le long de la chaîne peuvent se retrouver côte à côte et interagir fortement.
Pour modéliser cela sur un ordinateur quantique, nous avons besoin d'un Hamiltonien décrivant toutes ces interactions entre les acides aminés. Ensuite, nous pouvons prédire la structure finale en trouvant l'état qui minimisera l'énergie de notre Hamiltonien. Ici, nous allons nous concentrer sur la façon dont les chaînes d'acides aminés peuvent être modélisées sur un ordinateur quantique et comment nous pouvons obtenir les distances inter-acides aminés pour le calcul des énergies d'interaction. Avec cela, nous aurons rassemblé toutes les contributions nécessaires au Hamiltonien pour le simuler sur un ordinateur quantique.
Dans les vraies protéines, les acides aminés peuvent occuper un continuum de positions possibles. Cependant, nous allons utiliser une simplification et les contraindre à l'aide d'un modèle en réseau, où chaque acide aminé occupe un point sur une grille. Ici, les auteurs ont utilisé un réseau tétraédrique. Remarque rapide : ici, nous encodons la direction des arêtes, et non les nœuds eux-mêmes comme dans le problème Max-Cut. Chaque qubit représente un chemin possible d'un seul pas le long de la grille tétraédrique. Note que les sites adjacents ont été étiquetés A ou B en raison de leurs orientations différentes dans le réseau.
La chaîne protéique est représentée comme une série de virages ou de directions sur ce réseau. Chaque virage entre les acides aminés peut prendre l'une des quatre directions, correspondant aux arêtes du tétraèdre. Ces quatre virages possibles sont encodés en utilisant quatre qubits dans les états 0001, 0010, 0100 ou 1000.
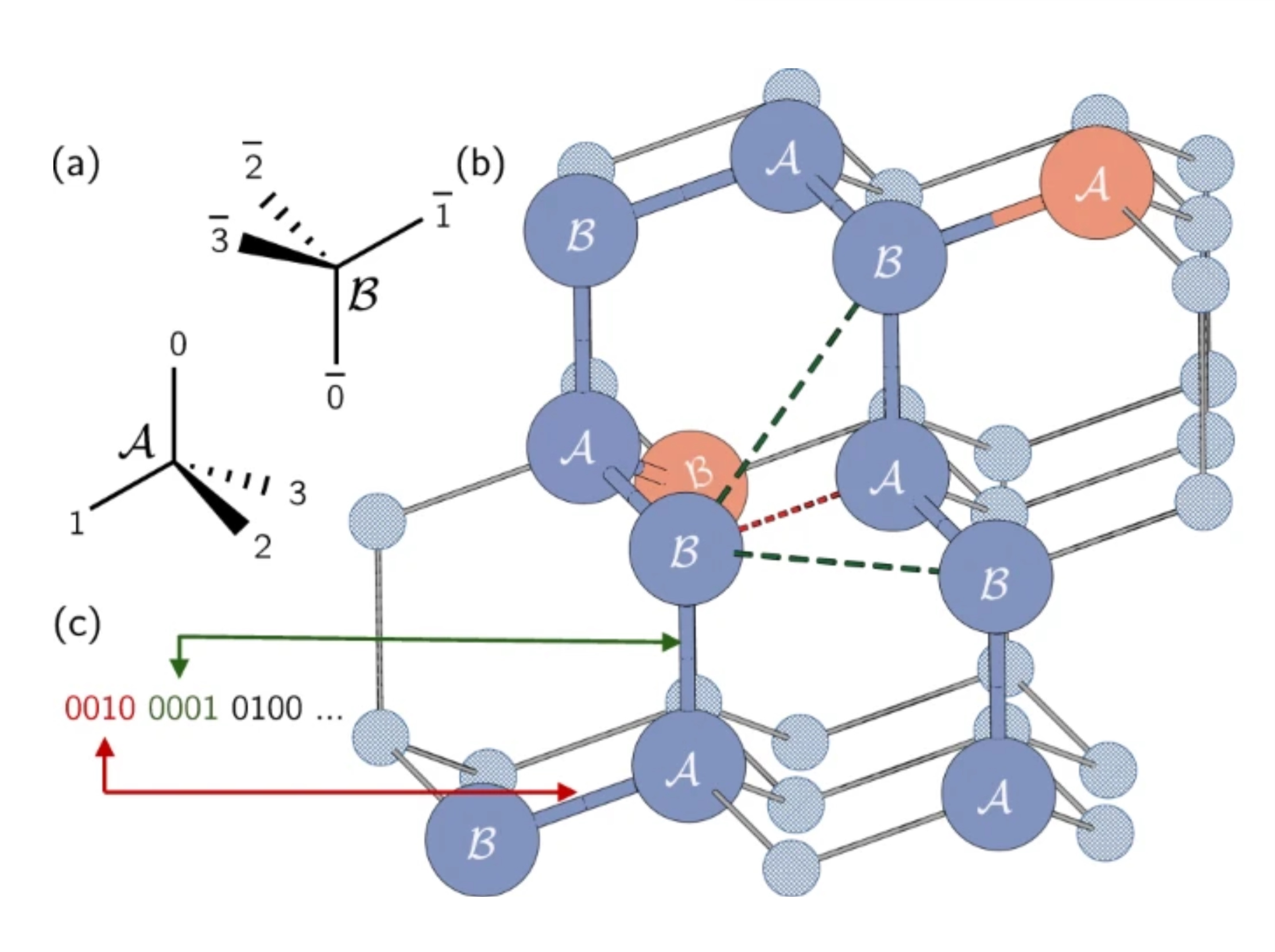
Regardons un exemple dans la figure ci-dessus. Plaçons notre premier acide aminé sur le point étiqueté « B » encerclé en rouge dans notre réseau tétraédrique. La direction du premier acide aminé vers le deuxième est arbitraire car le système peut toujours être tourné pour faire pointer cette arête dans n'importe quelle direction souhaitée. Ainsi, nous pouvons placer notre deuxième acide aminé sur le point en dessous du premier, étiqueté « A ». Ce n'est pas aussi facile à voir, mais le chemin du deuxième au troisième est également arbitraire. Les trois choix résulteraient en deux arêtes avec un angle d'environ 109,5 degrés entre elles. Choisir cette deuxième arête détermine simplement l'orientation de notre protéine dans l'espace. Ainsi, sans perte de généralité, nous pouvons choisir les deux premiers virages comme étant simplement 0001 et 0010.
Avec ces simplifications, la configuration de la chaîne d'acides aminés est donnée par cette expression :
Jusqu'ici, nous avons mis en correspondance les arêtes du tétraèdre avec des qubits, et notre circuit quantique sera un ansatz. Nous devons maintenant réfléchir à la façon d'encoder l'énergie du problème dans un Hamiltonien, de sorte que son état fondamental nous donne le modèle de repliement optimal.
Pour toute configuration donnée, il y aura une énergie associée due aux interactions entre les acides aminés. Ces interactions sont les plus fortes lorsque les deux acides aminés sont proches l'un de l'autre. De toute évidence, les acides aminés adjacents dans le squelette de la chaîne interagiront toujours entre eux. Mais parce que la protéine peut se tordre et se replier, d'autres paires d'acides aminés peuvent également interagir. Les acides aminés 10 et 20 pourraient par exemple se retrouver sur des sites adjacents après le repliement de la protéine. Nous avons donc besoin d'une formule pour décrire la distance entre les acides aminés et en utilisant l'information encodée sur les qubits de configuration. De cette façon, nous pouvons utiliser leur distance de séparation pour déterminer l'intensité de leur interaction.
D'abord, introduisons une fonction qui indique si une arête est utilisée ou non pour le virage au niveau de l'acide aminé . Ici, peut prendre l'une des quatre directions possibles. D'après la configuration avec laquelle nous avons débuté, nous pouvons écrire ces fonctions :
Nous pouvons ensuite définir une différence dans le nombre de virages étiquetés sur les réseaux A et B de l'indice à l'indice comme :
Pourquoi ferions-nous cela ? Eh bien, il s'avère qu'un virage de type d'un site A vers B est exactement annulé par un virage de type d'un site B vers A. Donc, pour connaître la distance entre l'acide aminé au site et celui au site , nous n'avons qu'à trouver la différence entre les pas effectués le long des arêtes depuis les sites A et les sites B. Puisque les sites A et B alternent nécessairement le long du squelette de la protéine, cela est capturé par le terme . Le même argument s'applique aux quatre types d'arêtes. Ainsi, la distance totale entre les acides aminés exprimée en pas tétraédriques peut être calculée par cette expression :
Mais comment obtenir le Hamiltonien à partir de cette longue équation pour la distance totale entre les acides aminés ? D'abord, nous pouvons convertir la distance en pas de réseau vers l'espace euclidien avec quelques calculs géométriques simples :
Ces distances seront ensuite utilisées pour calculer l'énergie de la configuration protéique. Selon nos objectifs, nous pourrions définir une distance seuil en dessous de laquelle nous considérons que la paire est en interaction, ou nous pourrions faire quelque chose de plus élaboré.
Cela ne saute peut-être pas aux yeux, mais nous avons en réalité terminé l'étape de correspondance en faisant cela. Les états des qubits indiquent le « virage » de la protéine à chaque site du réseau, et l'ensemble des virages détermine la distance entre toute paire d'acides aminés. Des paires de différentes espèces d'acides aminés ont des interactions différentes, certaines attractives, certaines répulsives. Si tu utilises la configuration et les distances pour simplement déterminer si les interactions connues entre acides aminés sont « activées » ou « désactivées », les intensités de celles-ci ont déjà été calculées et peuvent simplement être consultées dans un tableau comme celui-ci :
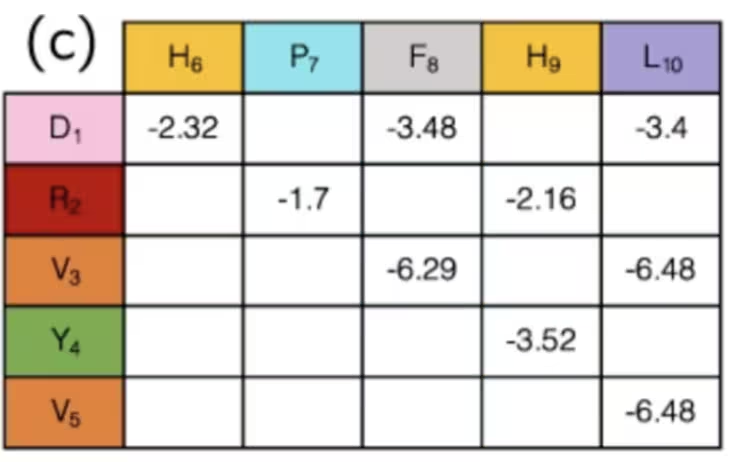
En résumé, dans cet exemple, les qubits sont utilisés pour marquer des pas sur un chemin le long d'un réseau, qui forment ensemble une chaîne d'acides aminés. En simulant comment ils se courbent et se replient, nous pouvons espérer trouver de meilleurs résultats dans la recherche médicale. Nous avons omis le calcul de quelques termes de ce Hamiltonien car ils étaient très spécifiques à ce problème, tandis que la définition des directions sur un réseau peut être appliquée plus généralement. Une fois que tu as un Hamiltonien général, tu voudras toujours le traduire en opérateurs de Pauli, qui sont natifs à l'ordinateur quantique. C'est ce dont nous allons parler ensuite.
Transformation de Jordan-Wigner
Voyons maintenant comment traduire un système de particules subatomiques en opérateurs de Pauli.
Les particules subatomiques se divisent en deux catégories : les bosons et les fermions. Les bosons, comme les photons ou le Higgs, obéissent à un certain ensemble de règles statistiques. Les fermions, comme les électrons ou les neutrinos, en obéissent un autre. La différence clé entre eux est que les bosons sont autorisés à occuper le même état — il n'y a aucune limite au nombre de bosons pouvant se trouver dans l'état fondamental ou tout état excité. Les fermions, en revanche, sont égoïstes — ils exigent que chaque particule ait son propre état quantique.
Les bosons ont également des spins entiers tandis que les fermions ont des spins demi-entiers, comme l'électron de spin 1/2, et des particules plus exotiques de spin 3/2. Pour décrire un système de particules, nous avons besoin d'une description de leur énergie. Concentrons-nous sur les fermions. Nous pouvons partir d'un Hamiltonien écrit en termes de ce que l'on appelle les opérateurs c. Ce sont essentiellement des objets mathématiques qui correspondent à la création ou à l'annihilation d'un fermion depuis un état dans le système. Ils sont souvent écrits comme et , où est l'opérateur qui crée un fermion dans l'état et est l'opérateur qui détruit un fermion dans l'état .
Mais rappelle-toi, les ordinateurs quantiques fonctionnent généralement dans une base de qubits avec des règles spécifiques pour représenter les systèmes fermioniques ; ils ne fonctionnent pas intrinsèquement dans le langage des opérateurs fermioniques. Pour combler cet écart, nous devons mettre en correspondance cette notation fermionique avec des opérateurs de Pauli, qui correspondent naturellement aux portes quantiques.
Il existe plusieurs transformations de ce type pour les fermions. Un choix courant est la transformation de Jordan-Wigner. Les encodages de Bravyi-Kitaev et d'encodage par parité sont des encodages fermioniques plus récents. Les opérateurs bosoniques peuvent être transformés en utilisant la transformation de Holstein-Primakoff ou en mappant directement les états de Fock vers une sous-base des modes bosoniques disponibles, parmi d'autres options. D'autres encodages font également l'objet de recherches actives. Pour l'instant, nous allons simplement nous concentrer sur la transformation de Jordan-Wigner.
La transformation de Jordan-Wigner implique de mettre en correspondance un seul fermion avec plusieurs qubits. Pourquoi ne peut-on pas simplement assigner un qubit pour représenter chaque électron ? Cela a à voir avec la distinguibilité des fermions identiques. Les qubits sont distinguables et les électrons ne le sont pas. Par exemple, nous pouvons facilement étiqueter et identifier les qubits individuels sur n'importe quel appareil. Mais l'indiscernabilité des électrons signifie que nous ne pouvons pas du tout les étiqueter. Ainsi, nous avons besoin d'étiqueter les opérateurs selon les orbitales occupées, comme 1s, 2p, 2p, etc., plutôt que les électrons spécifiques. Donc, chaque qubit joue approximativement le rôle d'une orbitale dans la molécule qui est soit occupée, soit inoccupée. Mais la façon dont nous faisons cela est un peu plus compliquée. La correspondance de Jordan-Wigner suit l'anti-symétrie et assure les statistiques correctes du système fermionique global. La correspondance de Jordan-Wigner exprime les opérateurs fermioniques en termes d'opérateurs de Pauli en utilisant ces relations :
La correspondance de Jordan-Wigner est conceptuellement simple en raison de la correspondance biunivoque entre orbitales et qubits. Il existe d'autres correspondances qui accomplissent un objectif similaire, notamment l'encodage par parité. Le calcul de la parité d'un état nécessite la prise en compte de plusieurs qubits. Dans l'encodage par parité (et certains autres), l'interprétation d'un qubit correspondant à une orbitale ne tient plus. Passons maintenant à travers un exemple simple. Supposons que nous voulions calculer l'interaction sur un seul qubit . Nous commençons par substituer nos définitions des opérateurs de création et d'annihilation.
Le commutateur . Ainsi, l'expression finale devient :
Nous avons donc réécrit avec succès une expression fermionique en termes d'opérateurs de Pauli que notre ordinateur quantique sera capable de comprendre.
Voyons rapidement comment nous implémenterions la correspondance de Jordan-Wigner dans Qiskit. Il est important de comprendre comment ces types de transformations fonctionnent, mais il serait peu pratique de les calculer manuellement à chaque fois — surtout pour des systèmes de grande taille. Heureusement, Qiskit nous facilite la tâche avec la fonction SparsePauliOp.
À un niveau élevé, les étapes sont :
- Utiliser la fonction
from_listde SparsePauliOp pour créer un opérateur identité correspondant à la taille de l'espace des paramètres à mettre en correspondance. - En suivant la définition des opérateurs de création et de destruction présentée précédemment, utiliser la fonction
from_listde SparsePauliOp pour définir les opérateurs de Pauli , , . Cela mettra en correspondance les opérateurs de création et d'annihilation fermioniques avec des opérateurs de spin de qubit, encodant le nombre d'occupation fermionique dans la base de calcul des qubits. - Générer le Hamiltonien souhaité dans la base de Pauli en appliquant les opérateurs ci-dessus aux orbitales d'intérêt. Cela correspond généralement à la création d'une matrice identité qui représente les orbitales du cœur (non-interagissantes), puis à l'application des opérateurs de création et d'annihilation à l'espace actif, avec des poids qui correspondent aux spécificités du problème en question.
Maintenant que nous comprenons pleinement le schéma de correspondance de Jordan-Wigner, examinons un exemple plus compliqué. Tu te souviens peut-être de l'article intitulé « Scalable circuits for Preparing Ground States on Digital Quantum Computers: The Schwinger Model Vacuum on 100 qubits » de la leçon précédente. Nous n'allons pas reprendre l'article en détail — nous allons simplement nous concentrer sur la correspondance de Jordan-Wigner, qui est utilisée pour exprimer le Hamiltonien de sites en réseau à sites pour le modèle de Schwinger en l'absence de champ électrique.
Ici, il est beaucoup plus difficile de dire précisément ce que représente un qubit dans ce modèle, car ce n'est que la collection de qubits ensemble qui forme quelque chose de physique, en l'occurrence un paquet d'ondes. À la place, tu peux considérer grossièrement les qubits comme des morceaux d'espace discrétisés. Ici, est le volume du réseau dans lequel chaque élément (maille élémentaire) comprend deux qubits. Les opérateurs fermioniques que nous avons vus dans la diapositive précédente décrivent l'amplitude de la fonction d'onde sur un site particulier. Notre Hamiltonien contient donc ces opérateurs de création et d'annihilation fermioniques. Nous utilisons donc la transformation de Jordan-Wigner pour mettre ces opérateurs en correspondance avec les opérateurs de Pauli.
où est l'opérateur de Pauli et est Une fois que nous avons un Hamiltonien écrit dans ce format, la partie difficile de l'étape de correspondance est terminée, et il peut maintenant être facilement écrit dans un circuit en opérateurs de Pauli.
Conclusion
Nous avons discuté de quatre exemples de la façon dont des techniques de correspondance spécifiques ont été utilisées récemment dans le domaine de l'informatique quantique, en commençant par les plus simples et en progressant jusqu'à l'application de la transformation de Jordan-Wigner à la dynamique des hadrons. Une grande partie de ce matériel était très technique, et si tu ne l'as pas vu auparavant, cela peut sembler très intimidant. Mais cela devient plus facile à mesure que tu passes du temps à pratiquer — c'est pourquoi ce cours s'appelle L'informatique quantique en pratique. Ce n'est pas quelque chose que tout le monde peut simplement saisir et mettre en œuvre dès le début — cela nécessite de l'étude, de la réflexion, et probablement des moments de frustration. Mais je t'encourage à accueillir cet inconfort et à explorer vraiment les questions qui surgissent au fil de ta progression. C'est le seul moyen d'apprendre.